9. Human population genetics
After this chapter, the students can name major resources of human genetic data and outline the major events in the history of modern humans.
1000 Genomes Project
The Human Genome Project in 1990-2003 aimed to sequence the entire euchromatic human genome as well as develop methods and ethical guidelines for its study. All its data were immediately freely available. The knowledge of the genome sequence was believed to help understand the mechanisms behind human diseases such as cancer. However, an equally important aim was to make “our” genome sequence open and free, and prevent commercial companies from patenting genes.
This was a real risk at the time: in early 1990s, researchers identified a gene, named as BRCA1, to be associated with breast cancer in high-risk families. The US genetic-testing company Myriad Genetics filed a patent on this gene and prevented anyone else from performing tests for mutations in that gene. The debate around this was long-lasting and ended only in 2013 by the decision of the US Supreme Court to disallow the patenting of naturally occurring DNA sequences.
The Human Genome Project provided a free and open access to the genome sequence, but it became soon evident that a single genome represents poorly the natural genetic variation among humans. Genetic variation was studied but often in medical context and the data were not publicly available. Furthermore, a great majority of studies focused on individuals of European origin.
The 1000 Genomes Project (1KGP), taken place from January 2008 to 2015, was the answer to this and set to sequence the genomes of at least 1000 individuals of diverse ethnic background. Unlike most genomes so far, 1KGP focused on anonymous healthy individuals and aimed to identify common genetic variants with frequencies of at least 1% in the populations studied. The project also developed guidelines on ethical considerations for investigators doing sampling as well as bioinformatic methods to investigate and share the data.
The populations included in 1KGP are shown below. Geneticist Leena Peltonen-Palotie was central in convincing the steering group that the Finnish population, due to its peculiar background, would be a valuable addition for medical genetics and should be included among the study populations.
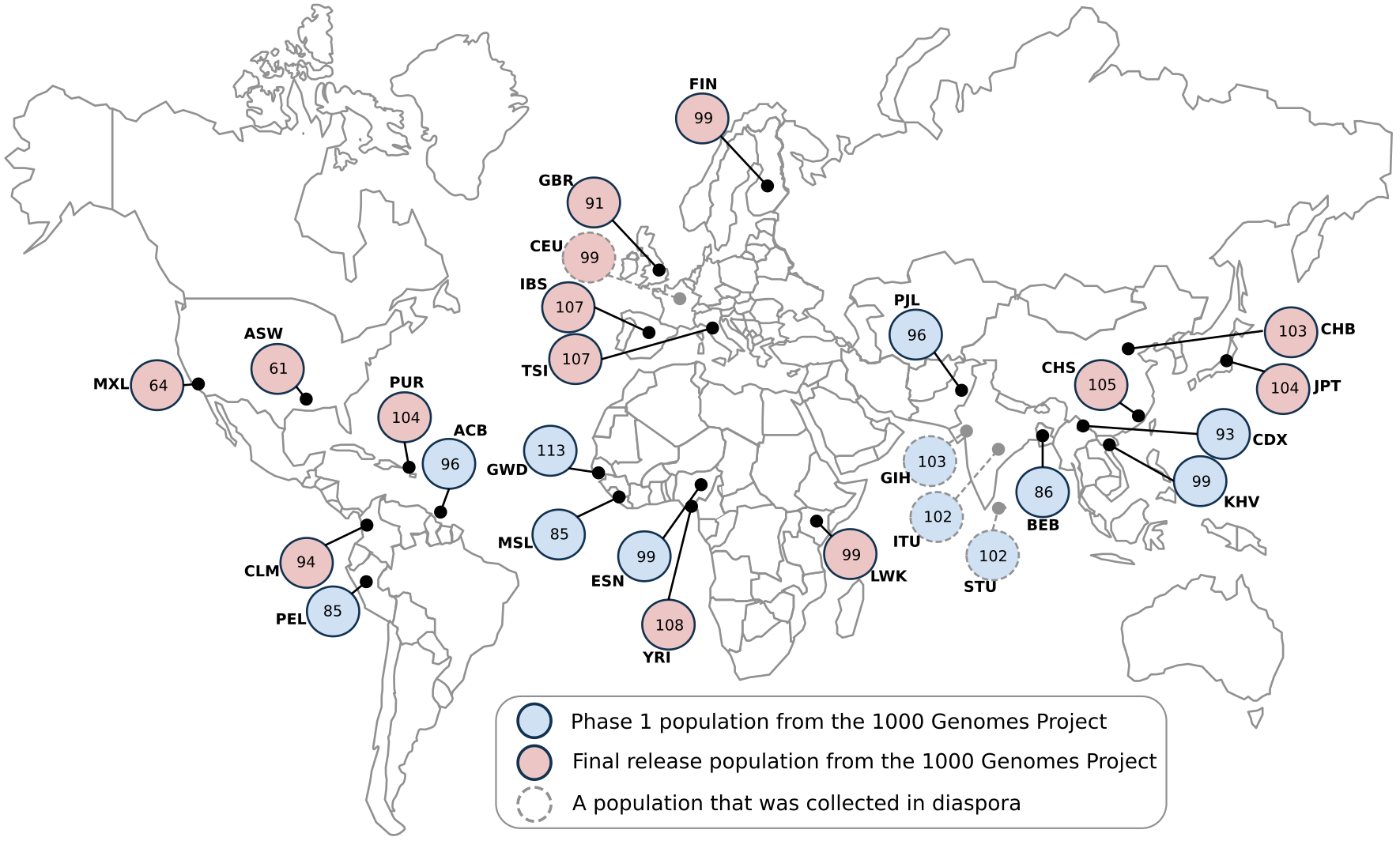
| ID | Population |
|---|---|
| ASW | African Ancestry in Southwestern USA |
| ACB | African Caribbean in Barbados |
| BEB | Bengali in Bangladesh |
| GBR | British from England and Scotland |
| CDX | Chinese Dai in Xishuangbanna, China |
| CLM | Colombian in Medellín, Colombia |
| ESN | Esan in Nigeria |
| FIN | Finnish in Finland |
| GWD | Gambian in Western Division – Mandinka |
| GIH | Gujarati Indians in Houston, Texas, United States |
| CHB | Han Chinese in Beijing, China |
| CHS | Han Chinese South, China |
| IBS | Iberian populations in Spain |
| ITU | Indian Telugu in the U.K. |
| JPT | Japanese in Tokyo, Japan |
| KHV | Kinh in Ho Chi Minh City, Vietnam |
| LWK | Luhya in Webuye, Kenya |
| MSL | Mende in Sierra Leone |
| MXL | Mexican Ancestry in Los Angeles, California, United States |
| PEL | Peruvian in Lima, Peru |
| PUR | Puerto Rican in Puerto Rico |
| PJL | Punjabi in Lahore, Pakistan |
| STU | Sri Lankan Tamil in the U.K. |
| TSI | Toscani in Italia |
| YRI | Yoruba in Ibadan, Nigeria |
| CEU | Utah residents with Northern and Western European ancestry from the CEPH collection |
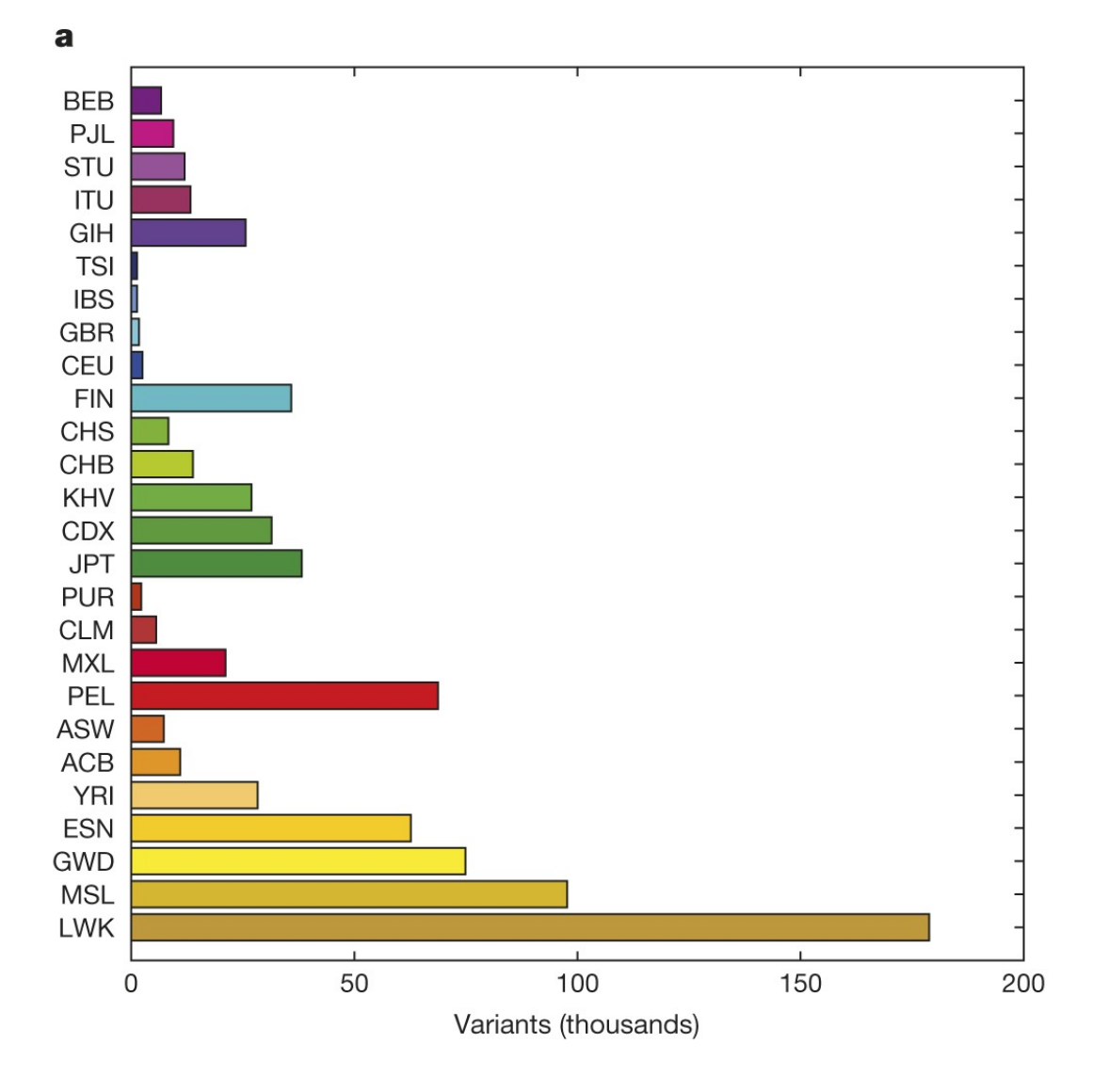
The findings of 1KGP are summarised in the final article. Some of the findings should look familiar by now:
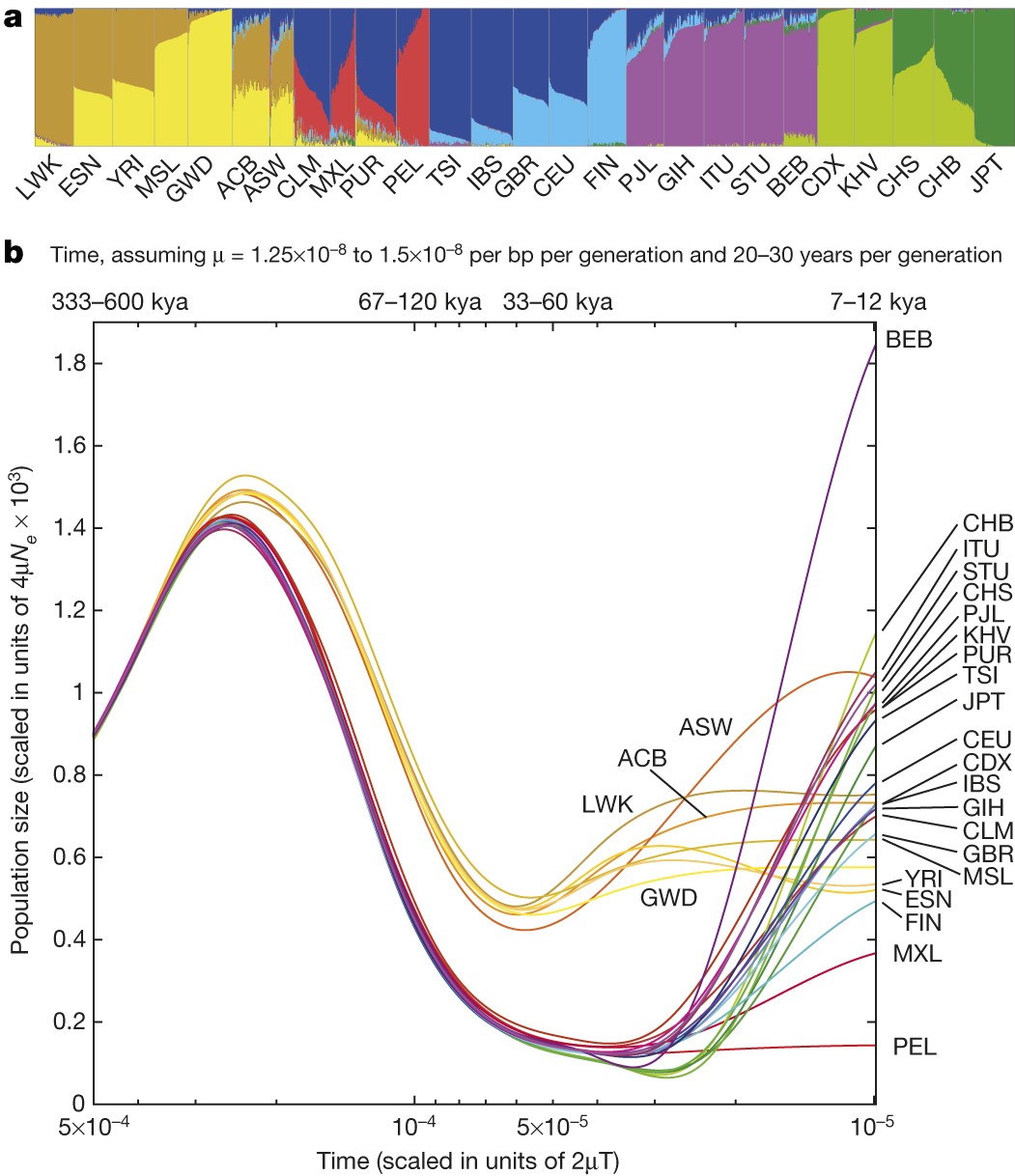
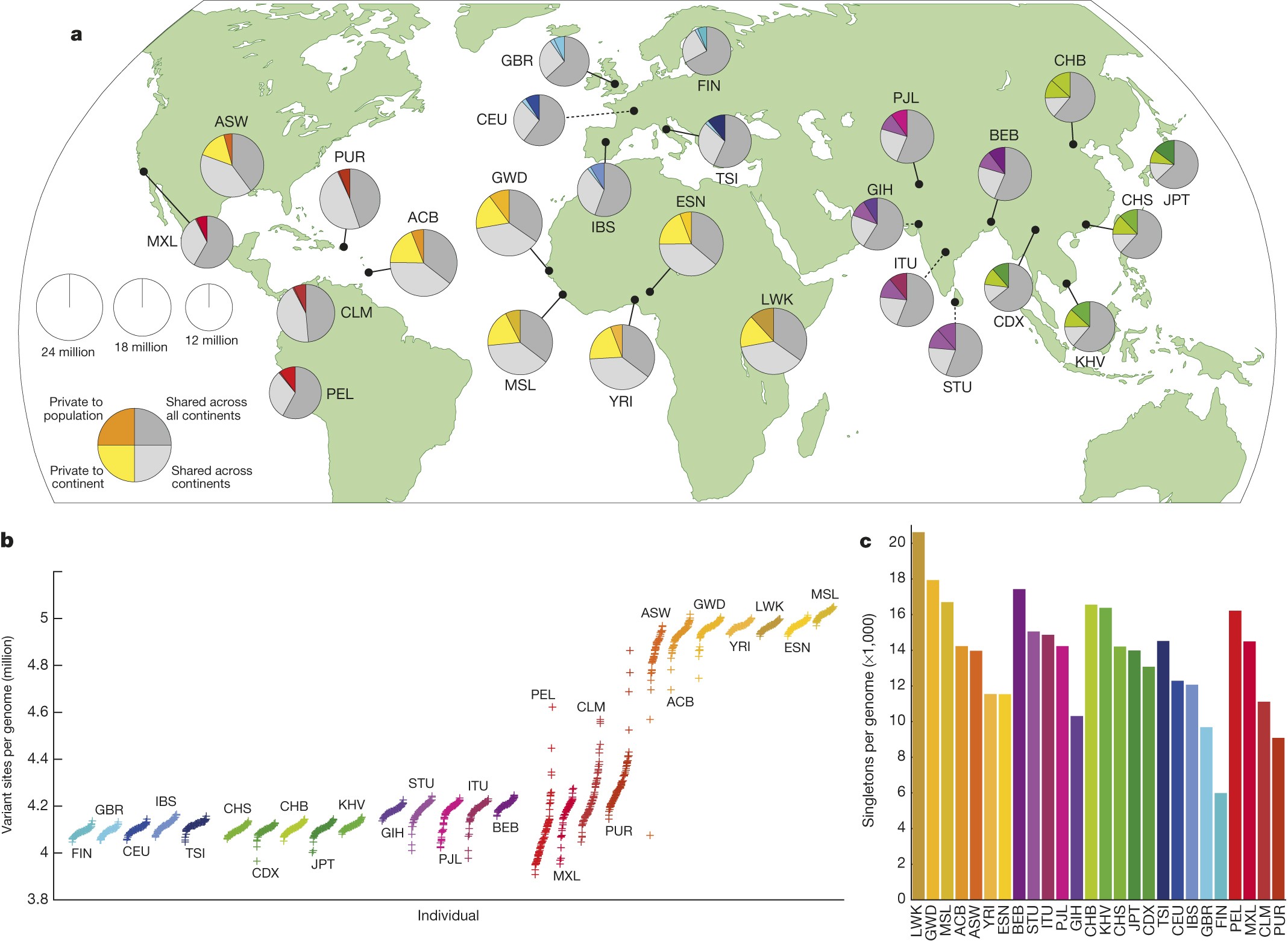
Leena Peltonen’s insights were confirmed (unfortunately, after her premature demise) and Finns were found to be enriched with variants that are rare (<0.5%) within the global sample but common (>5%) within a population.
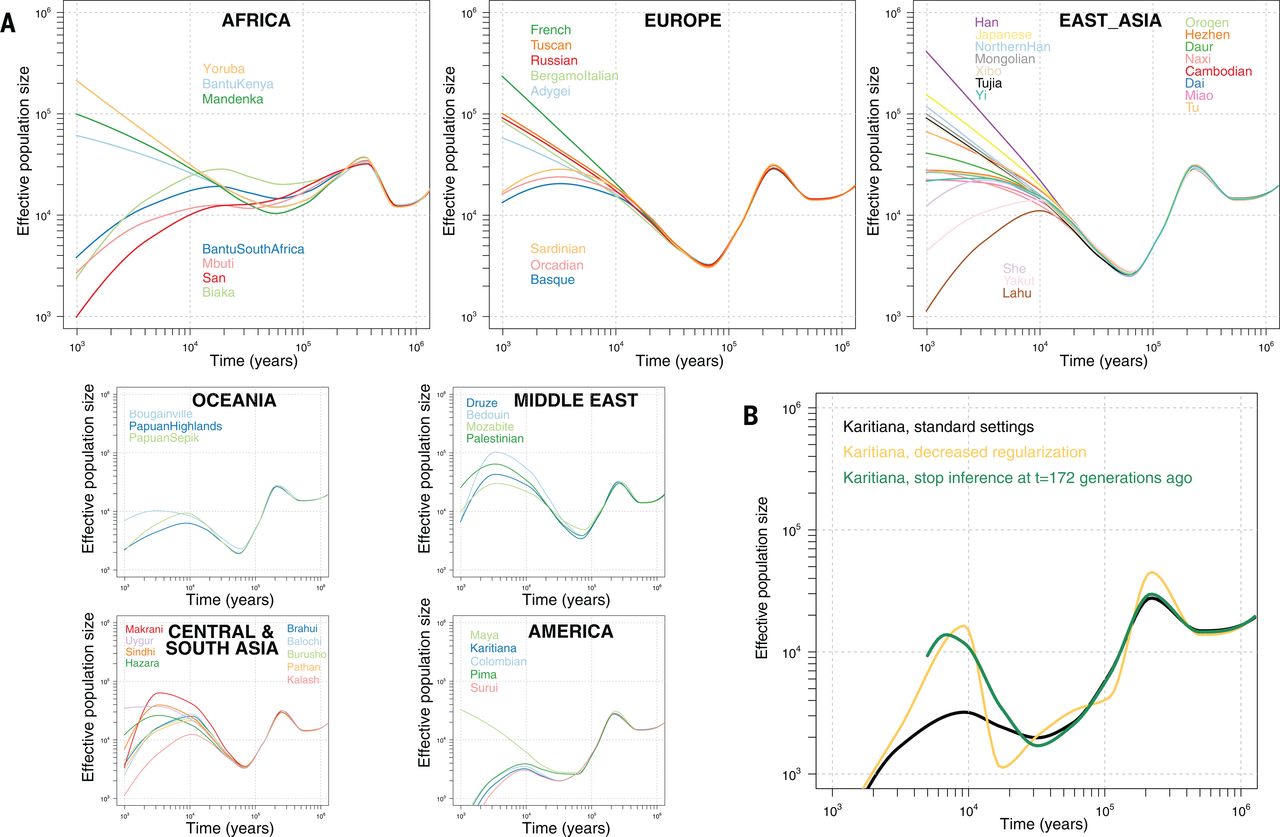
Other human genetic diversity projects
The 1000 Genomes Project was poorly named and, as its funding ended and the number of genomes greatly exceeded one thousand, a new entity, the International Genome Sample Resource (IGSR), was created to host and expand on the data set. There are other projects similarly aiming to understand the human genetic diversity, two significant ones being Human Genome Diversity Project (HGDP) and Simons Genome Diversity Project (SGDP).
Also the findings of HGDP have a familiar look:
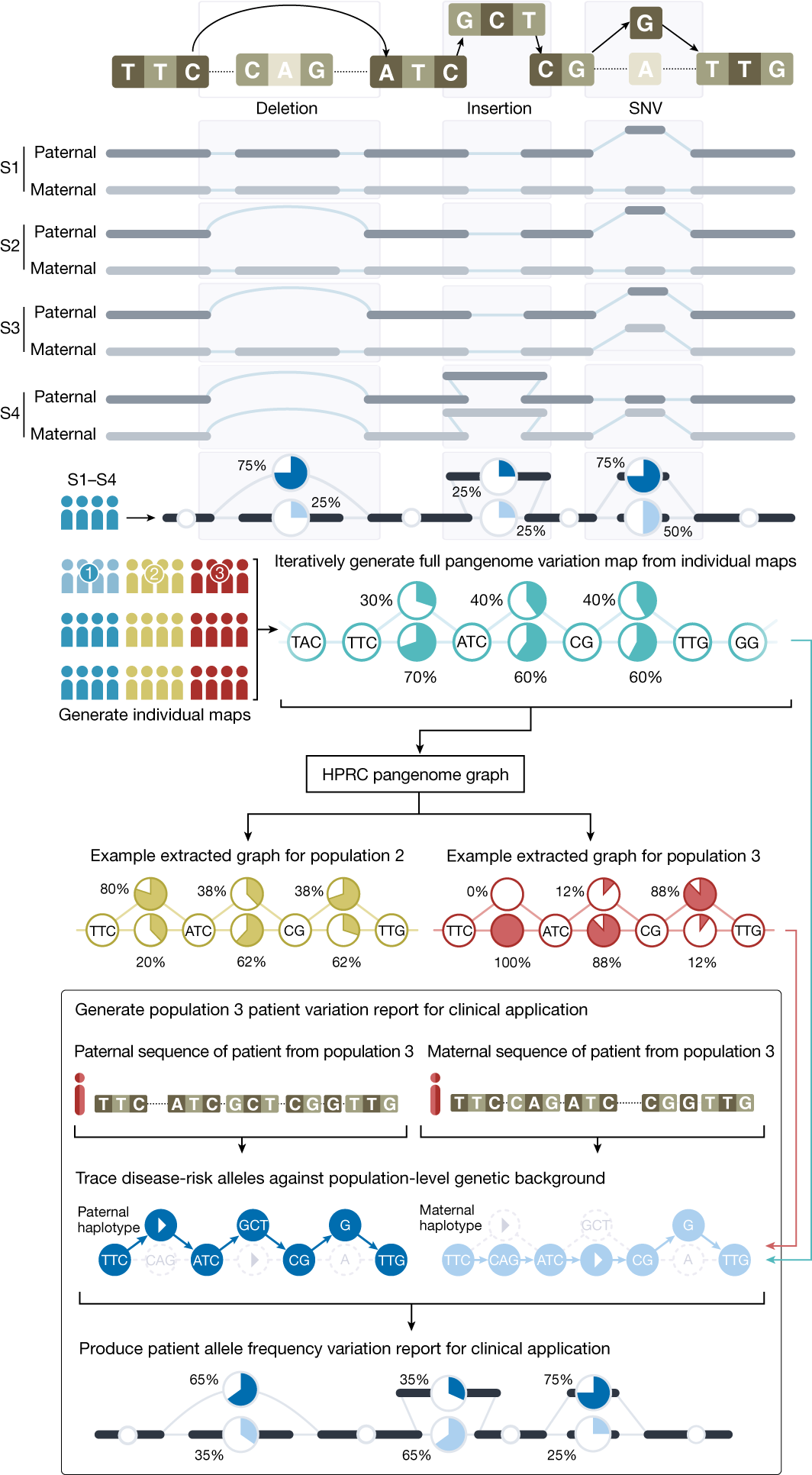
While these projects have also studied indels and copy-number variation, the data are generated with short-read sequencing and mapped against the standard human reference genome. This approach cannot reliably detect large structural changes. To counter that, the US National Human Genome Research Institute started a project to sequence and indepedently assemble genomes from individuals from diverse populations. The Human Pangenome Reference Consortium (HPRC) provides the resulting data in a raw format, mapped to a reference, as assemblies and in the form of a pangenome. Much of the data is openly available. A pangenome is no more a linear haploid sequence but a graph, and the proposed output of the project is very different from anything else so far:
A slightly different project is the Genome in a Bottle project by the US National Institute of Standards and Technology (NIST). Instead of diversity, the project helps to verify that the different sequencing platforms and sequence-processing software produce consistent results. The project shares results on a small number of samples that are repeatedly sequenced and analysed by the different organisations. The sample that is probably most studied in the world is NA12878, a mother of the CEPH family 1463:
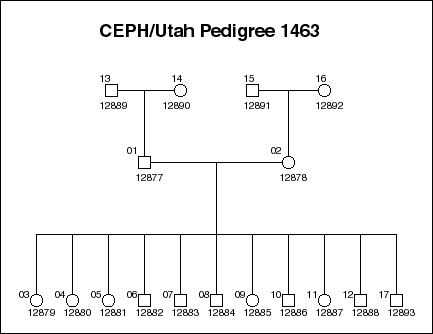
In addition to NA12878, her husband, their parents and the 11 children are available as lymphoblastoid cell lines for anyone to buy. Some members of the family are among the CEU samples studied in 1KGP, and the whole family were sequenced by the company Illumina.
CEU stands for Caucasians of European origin from Utah. In other contexts they are called Mormons.
CEU have been among the study populations from the very early genetic diversity projects. A central reason for this is that Mormons are extremely interested in their ancestry and family trees, and thus willing to help understanding their own past using modern methods. Mormons believe that families will join in the eternal afterlife. A more controversial is their belief that “temple rites” can also be performed for dead people by living proxies, in other words, dead people can be baptised to become Mormons as long as their identities are known. This has raised some controversy, especially as Mormons have posthumously baptised Jews who died in the Holocaust.
Genetic diversity of ancient humans
The Allen Ancient DNA Resource (AADR) provides curated genotypes of present-day and ancient DNA data. In contrast to other resources, AADR is basically a set of files which the studies of Reich lab are based on. AADR may be difficult to find and cumbersome to use, and gives an impression that is set up only to fulfil the openness criteria by the publishers.
Old versions of AADR are available under Reich labs’s website; the latest and future datasets should be under the Harvard University Dataverse.
Modern humans
One of the impressive findings of the early analyses of large human data, in this case 3,000 Europeans genotyped (not sequenced!) at half a million variable DNA sites, showed that genetic diversity mirrors the geography within Europe.
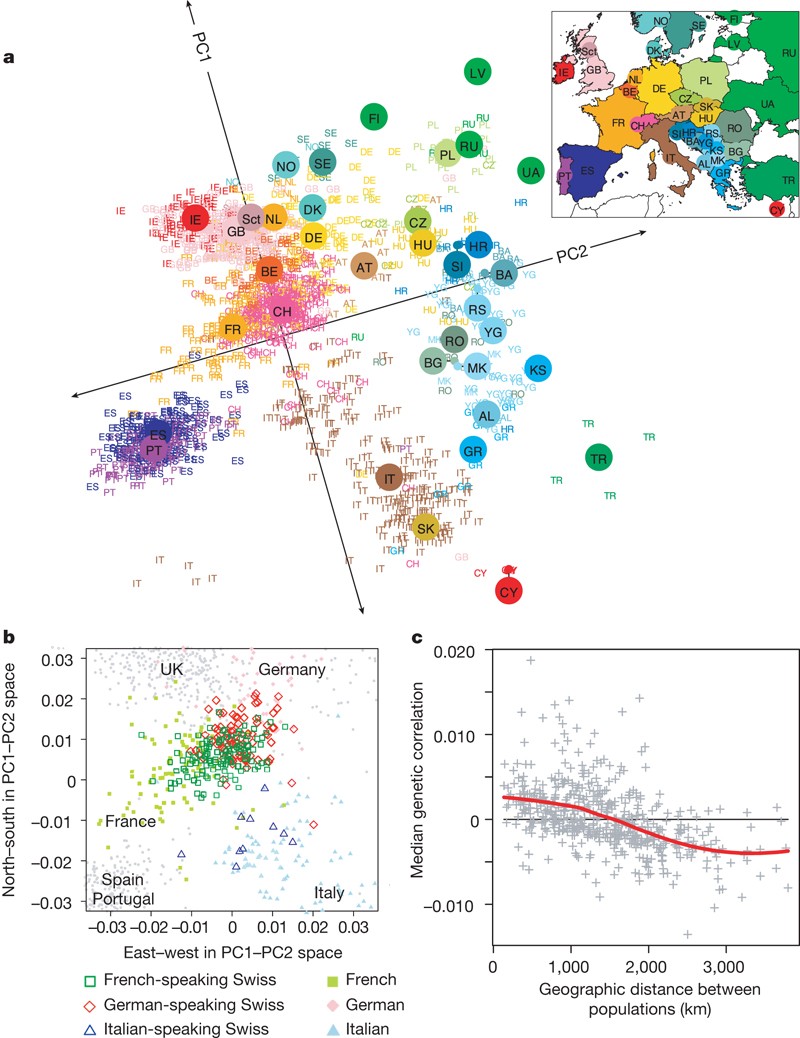
A downside of genotyping chips was that they could only detect the variants included in the test set and were thus not able to identify new variation from outside the populations used in the chip design. The early chips were heavily biased towards variation common among Europeans (there were valid medical applications with that design!) and did poorly in studies of global genetic diversity. The situation changed with the lowering costs of DNA sequencing. Probably even more dramatic was the development in analysis of ancient samples and ability to reconstruct and study the genetic diversity in past time points.
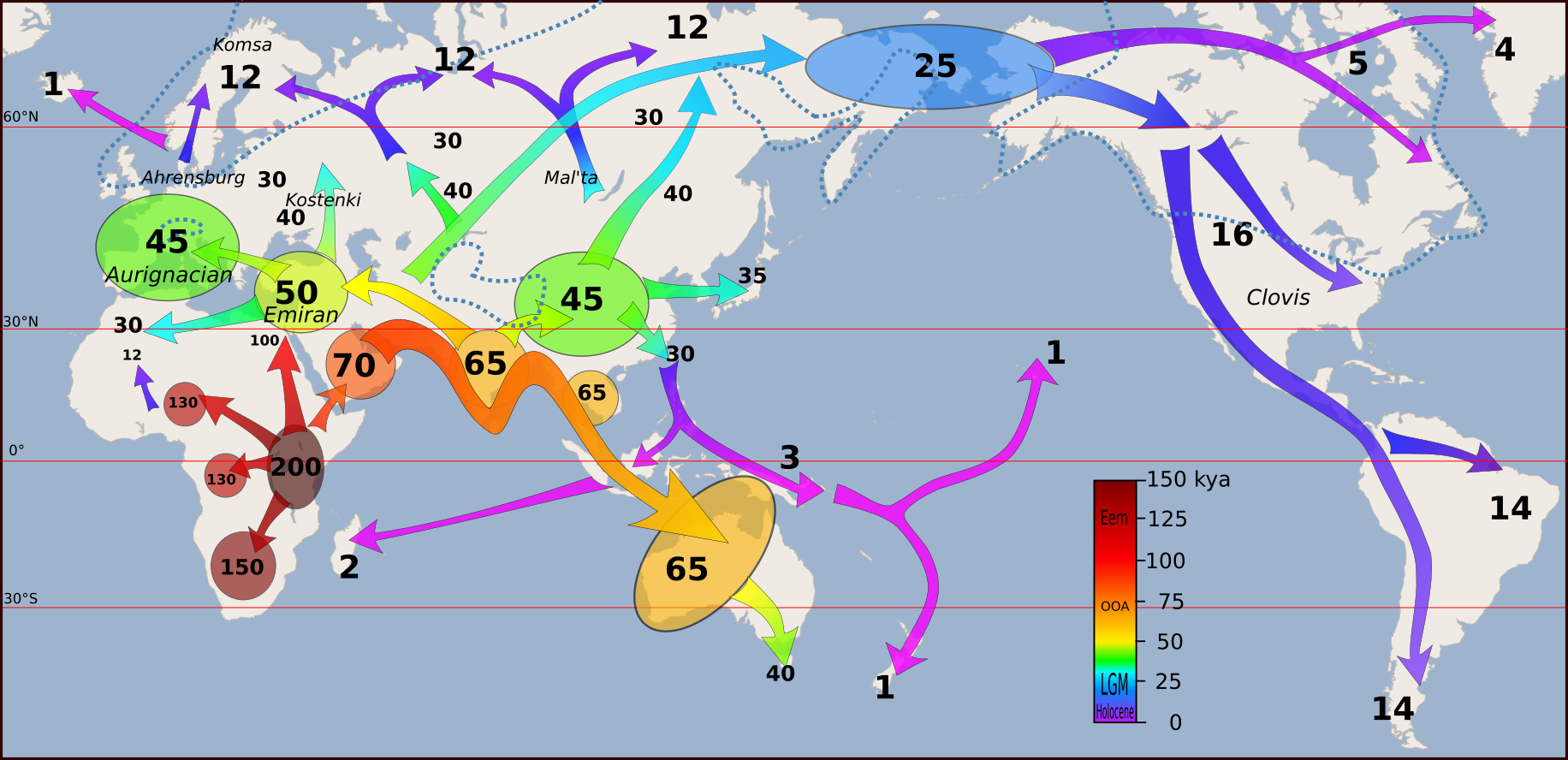
Based on the analyses of modern and ancient samples, the model of modern humans’ migration and colonisation of the world is following:
Admixture among archaic and modern humans
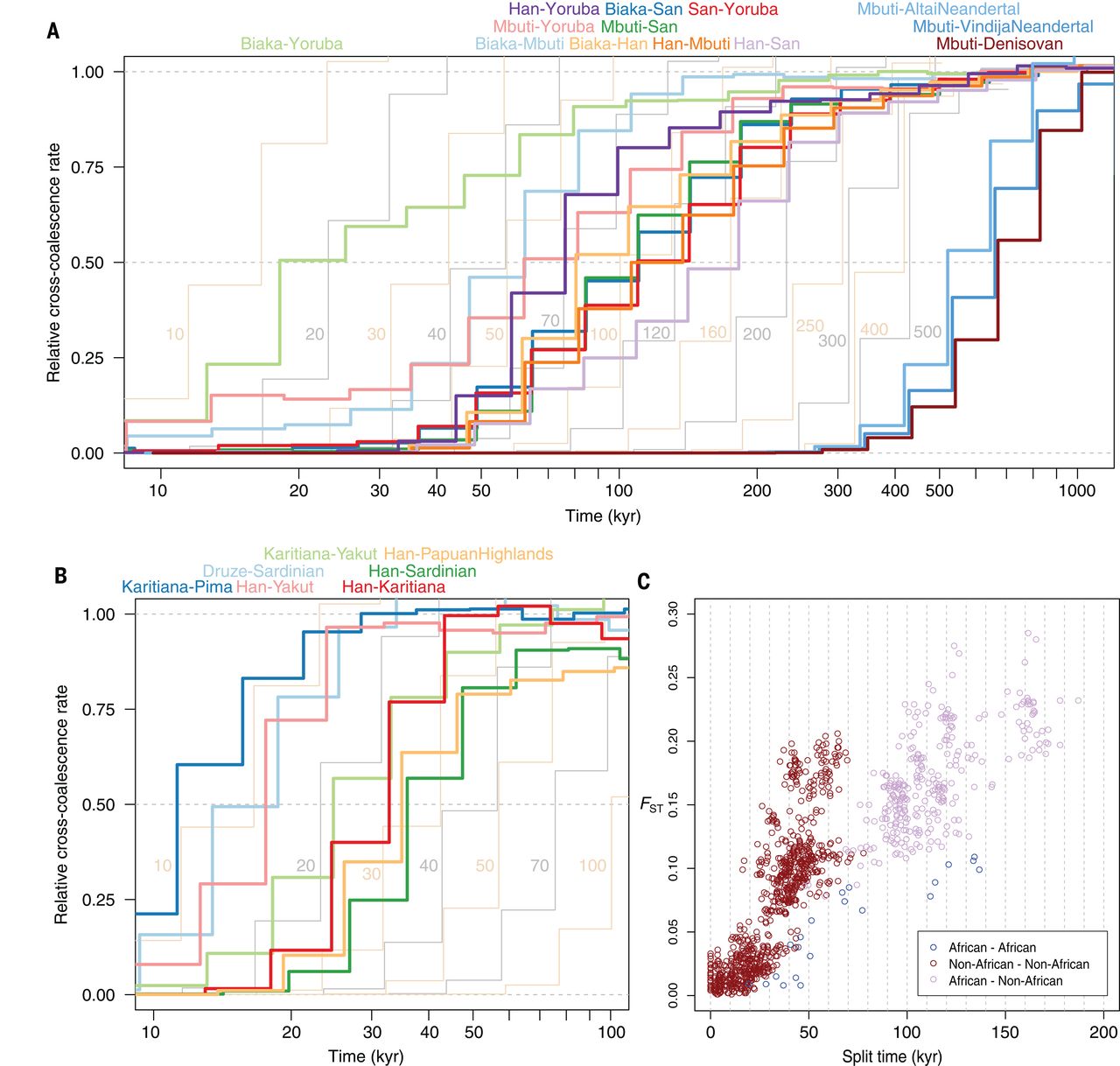
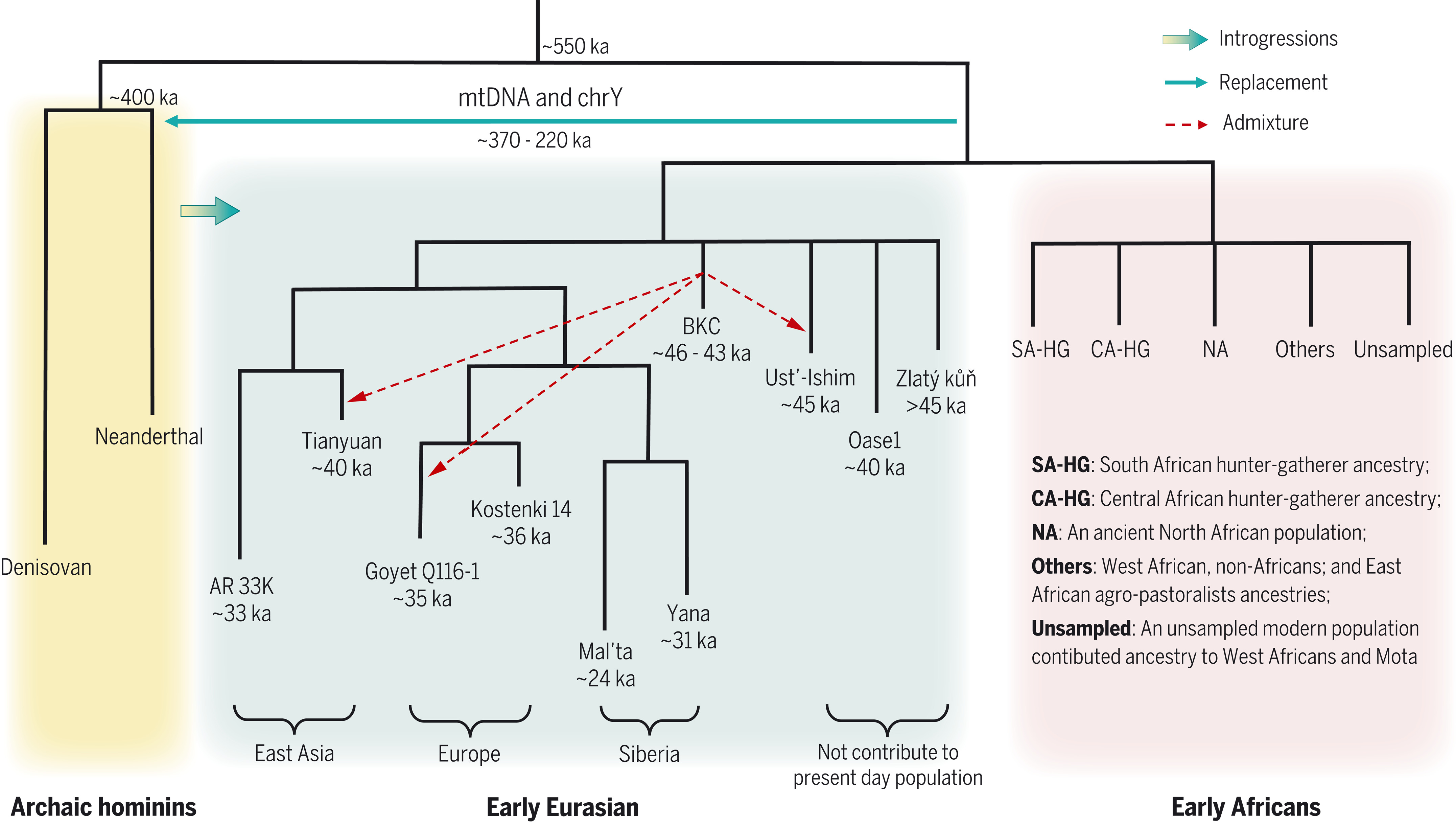
The human history is reviewed in Liu et al. (2021). Archaic humans are estimated to have separated from modern humans ~550 ka, Neanderthals and Denisovans splitting from each other ~400 ka.
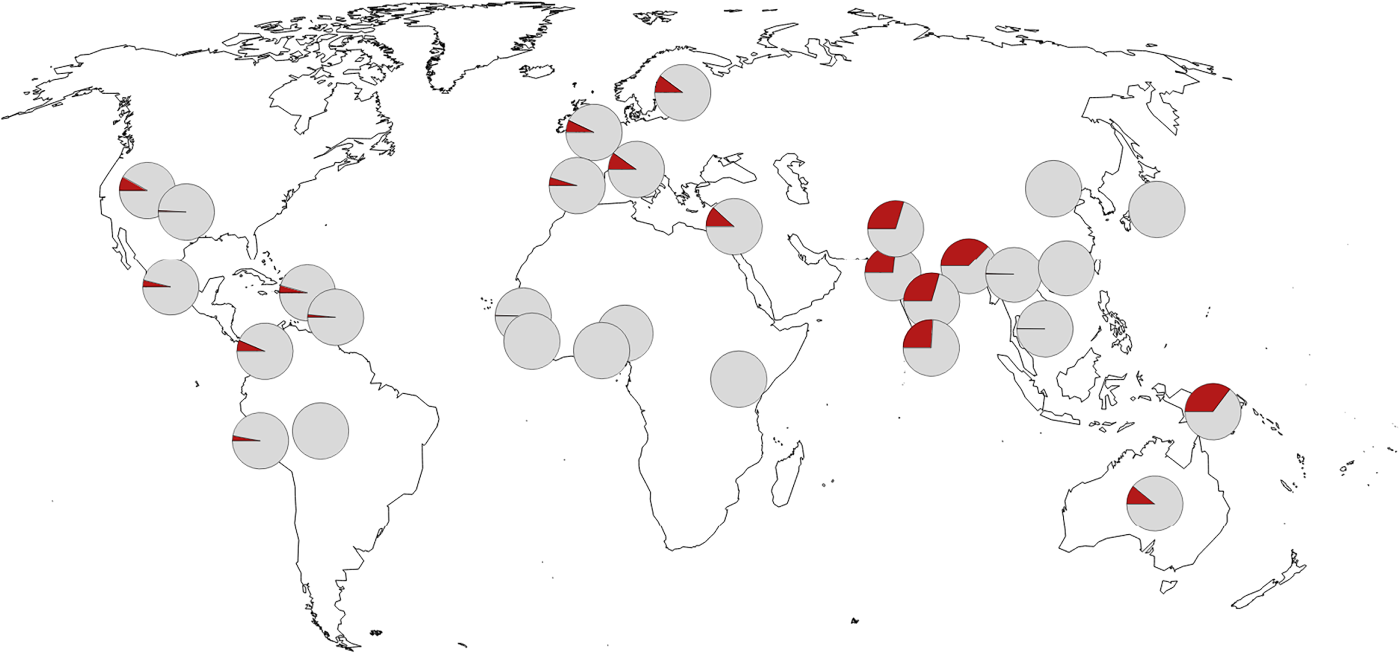
There was admixture between Neanderthal and Denisovan lineages and introgression to (and from) modern humans. All present-day non-Africans contain Neanderthal-derived ancestry, East Asian genomes containing a higher amounts (2.3 to 2.6%) than those of western Eurasians (1.8 to 2.4%). Very little Denisovan ancestry is found in western Eurasians while in Southeast Asia, the Denisovan ancestry proportions can be higher than Neanderthal ancestry, especially among Papua New Guineans and Oceanian people.
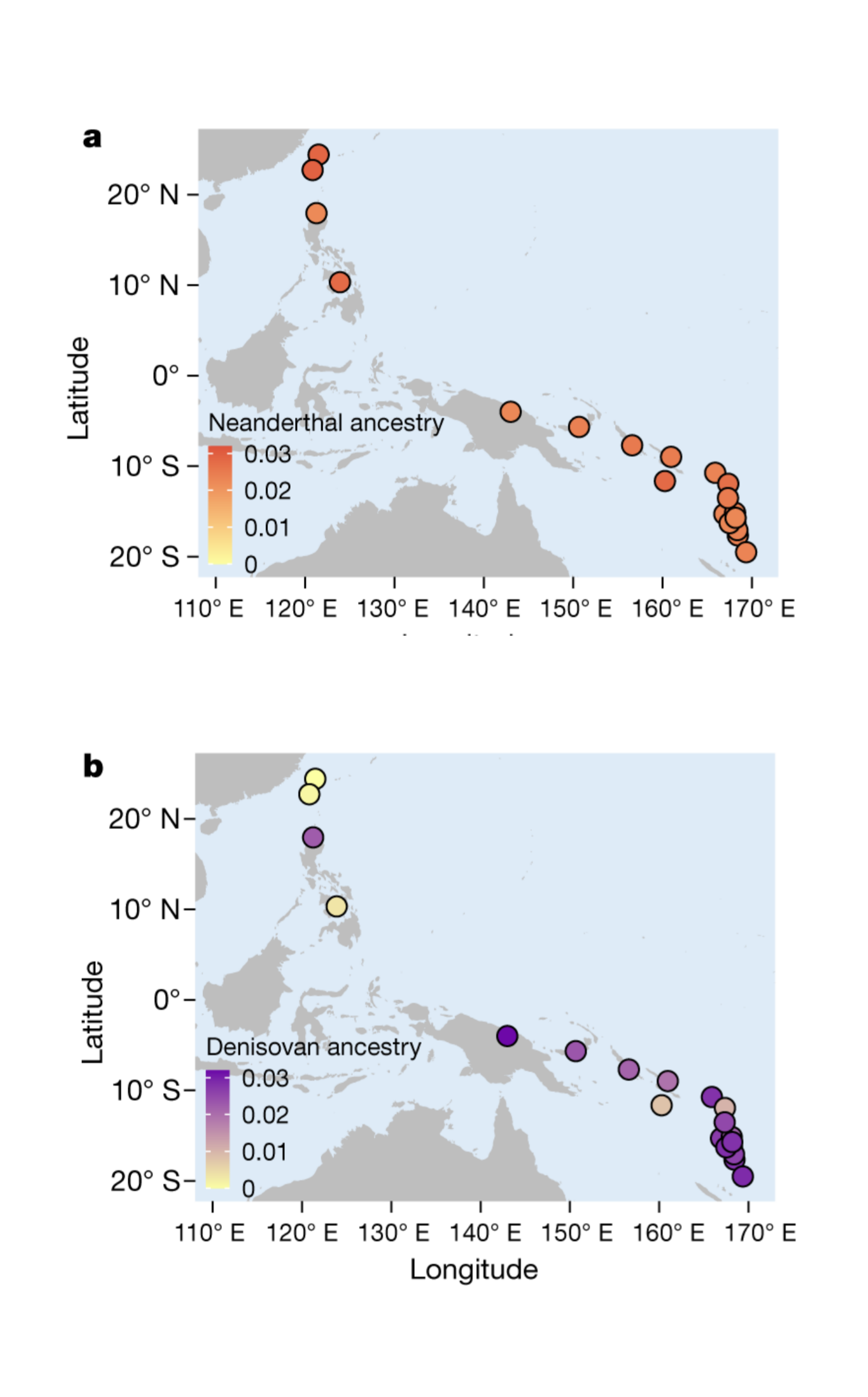
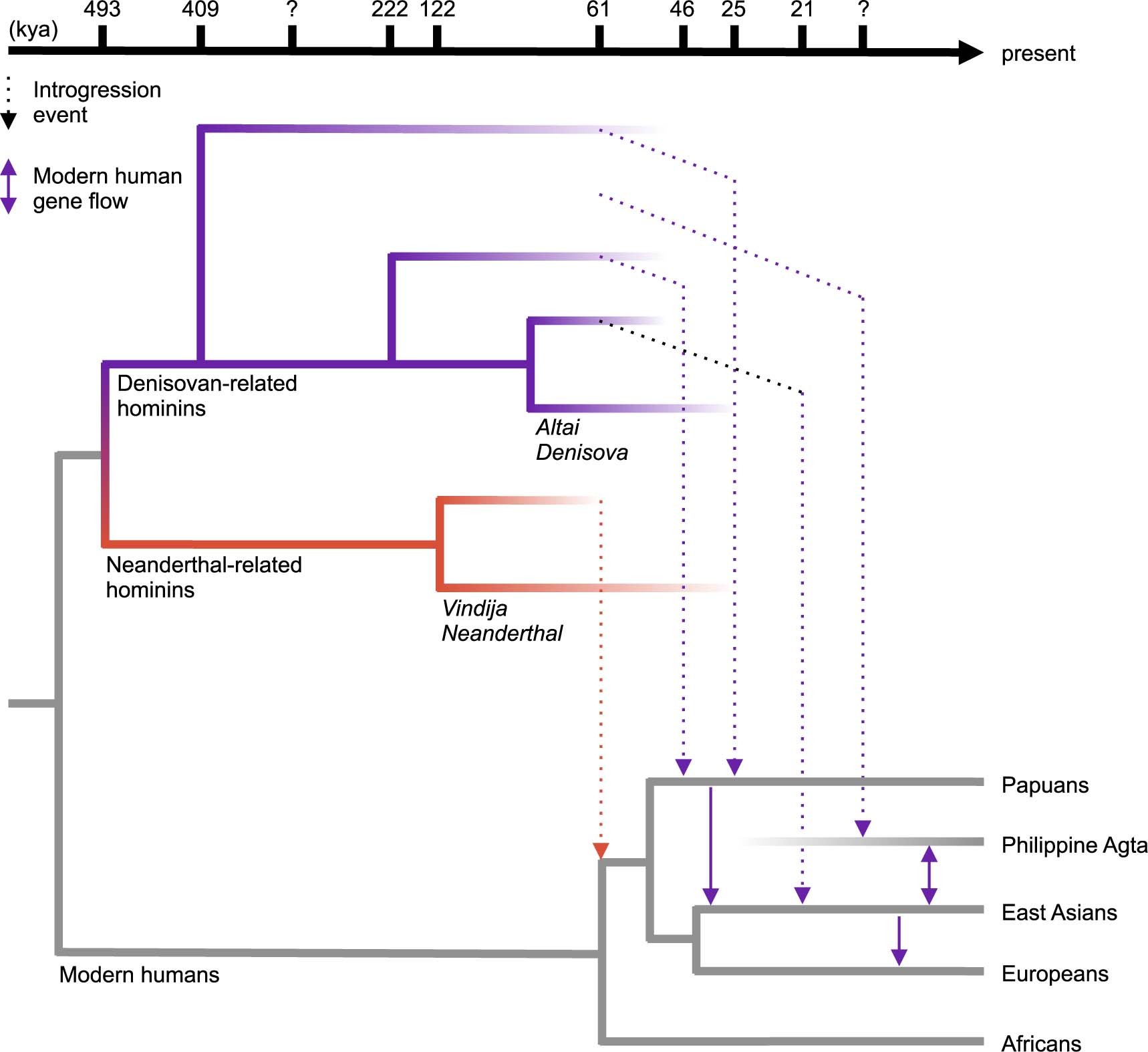
The varying fractions of archaic ancestry indicate that multiple pulses of introgression from Neanderthals and Denisovans to modern humans, occurring in different places across the Eurasian continent. Native Americans derive their ancestry from East Asians and Ancient North Eurasians from Siberia, thus carrying both Neanderthal and Denisovan variants.
Although Neanderthal-derived ancestry makes less than 3% of each modern human’s genome, the bits carried differ among individuals and, taken together, they span approximately 20% of the Neanderthal genome. Introgression from archaic humans is thought to have been generally negative and selection has removed material in gene regions. A recent study found that a major genetic risk factor for severe COVID-19 has been inherited from Neanderthals:
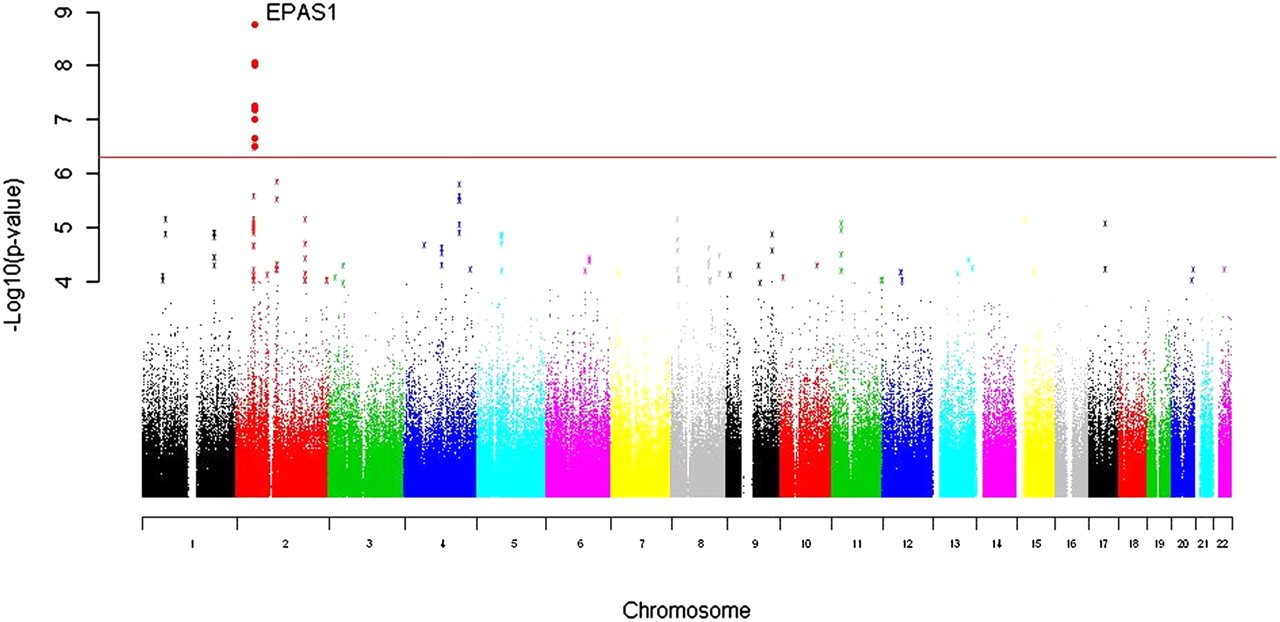
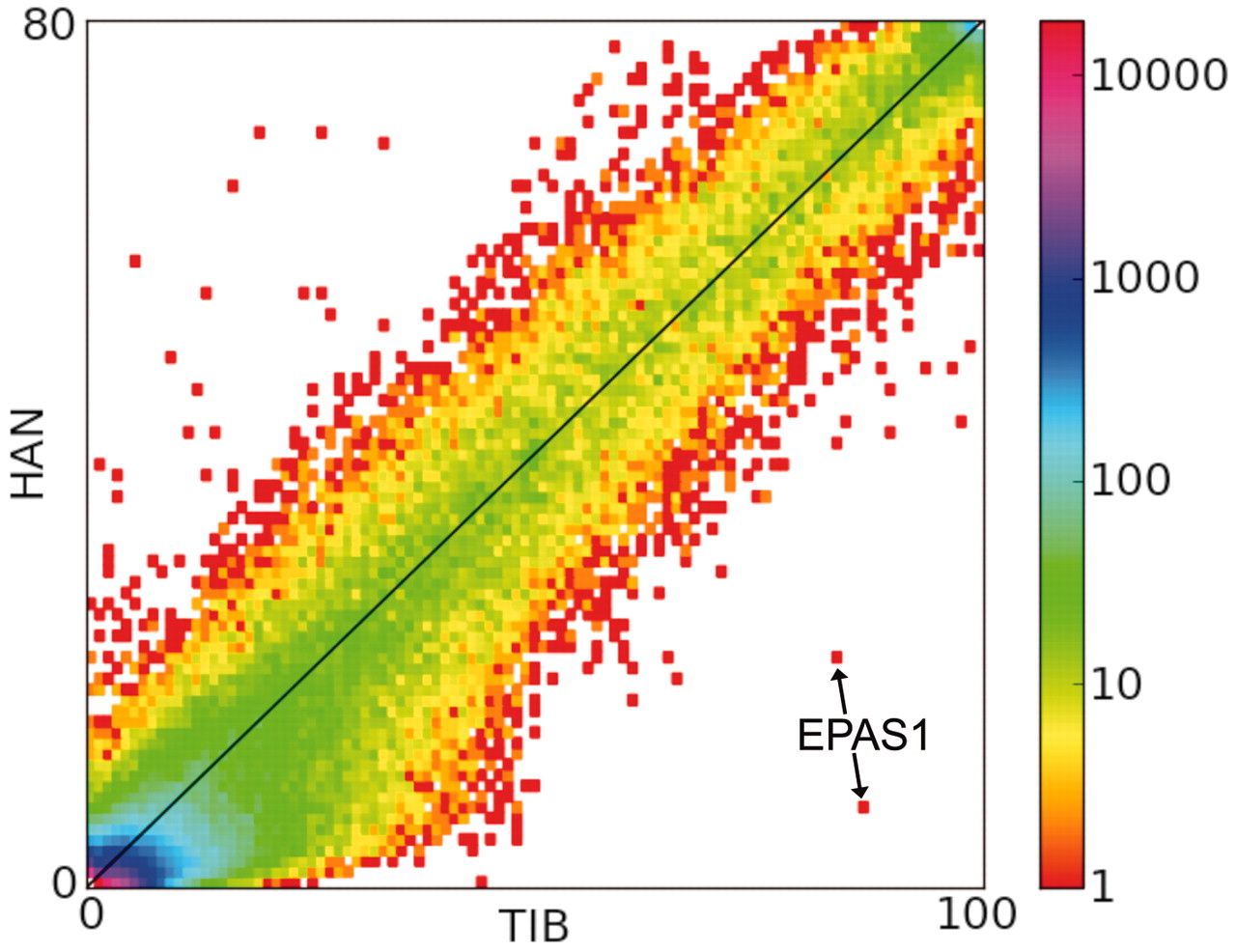
On the other hand, there are indications of introgression from Denisovans to Oceanians that may have helped to adapt into the harsh island conditions. One of the most striking examples of adaptive introgression from Denisovans to modern humans are the changes in the hypoxia pathway gene, EPAS1, in Tibetans where the Denisovan-derived haplotype is associated with differences in haemoglobin concentration at high altitude:

Modern humans in Europe
The early history of Europeans is reviewed in Lazaridis (2018).
The earliest humans in Europe were Neanderthals, inhabiting the area ~430–39 kya. Neanderthals were genetically differentiated from other archaic people, including at least Denisovans in Siberia and possibly elsewhere, and modern humans in Africa. The worldwide expansion of modern humans started ~60 ka and there are findings in Europe of modern humans that are up to ~45 ky old. However, several of the early lineages have not contributed to the ancestry of modern Europeans, indicating that they were dead-ends and went extinct.
The earliest samples from western Europe clearly sharing ancestry with Europeans but not East Asians is from Belgium and ~35–34 kya. The first major phase, Western European hunter-gatherers (WHG), appeared during a warm period ~15 kya and a WHG component is seen in the ancestry of modern Europeans. The Last Glacial Maximum, the ice age, had a major impact on all life in the higher altitudes.
After the LGM, the early Europeans went through a series of rapid expansions, migrations, interactions, and replacements. ~8 kya, the first farmers spread from Anatolia via southeastern Europe and admixed with the existing people. The Anatolian farmer ancestry reached Scandinavia and Iberia, but also some WHG populations persisted. In places, local WHG ancestry populations expanded again.
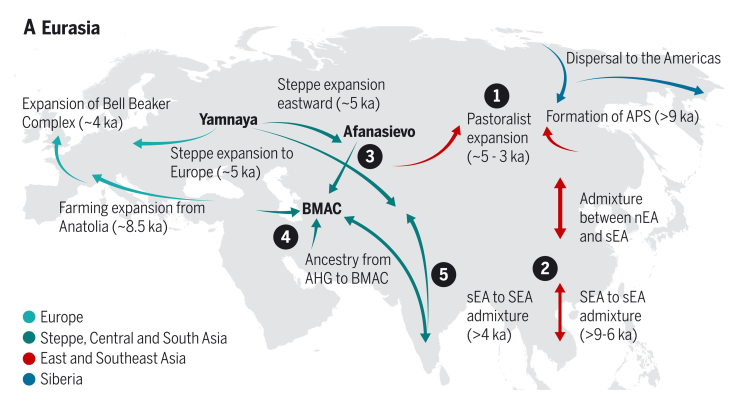
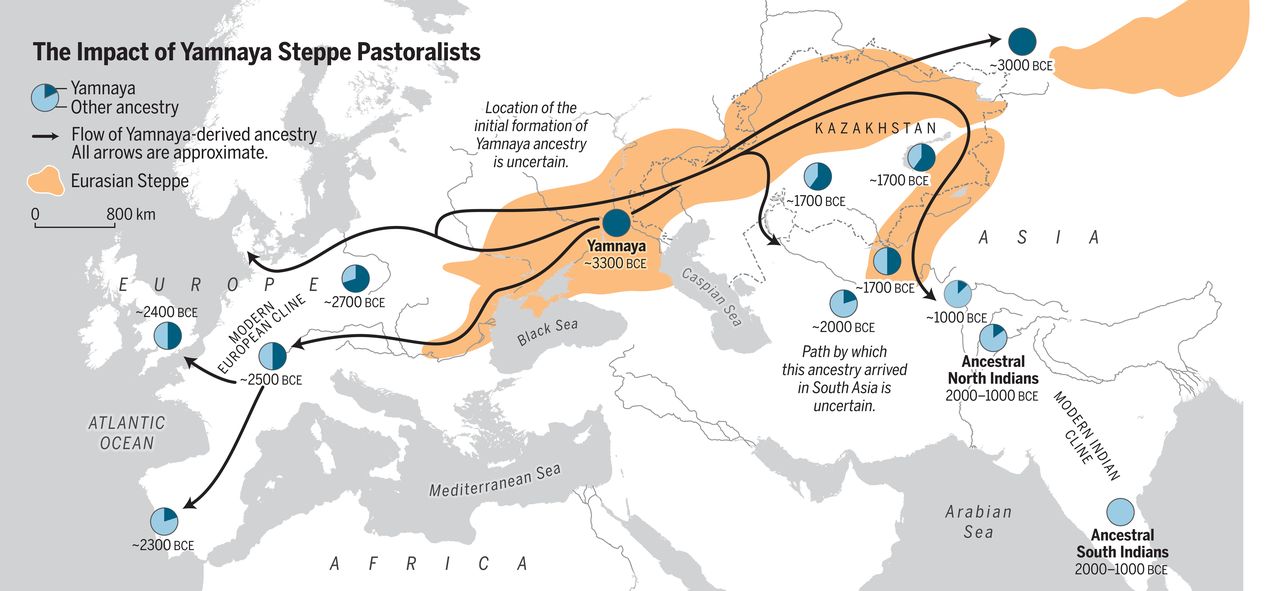
A central part of Eurasian human history are the steppe areas, open grasslands spanning with little breaks from Hungary to eastern China. A dominant population from the Pontic-Caspian steppe are the Yamnaya pastoralists that spread western Europe ~5 kya (and elsewhere). The Yamnaya expansion did not only introduce novel culture in western and central Europe but is believed to have given rise to the largest language group, Indo-European languages.
The three major genetic origins of modern Europeans are the European hunter-gatherers, Anatolian farmers and the steppe pastoralists, and most Europeans can be modelled as a mixture of these three ancestral populations. The ancestry proportions vary and include some exceptional populations such as Sardinians that are nearly clean descendants of the Neolithic Anatolian farmers.
Genetic history of Finns
The genetic history of Saami and Finns is studied in Lamnidis et al. (2018).
In north-eastern Europe, we have a few populations that do not speak Indo-European languages, notably Finns and Saami who speak Finno-Ugric languages of the Uralic language family. There has been human presence in Finland for 9000 ky, but the Finno-Ugric branch did not diverge from other Uralic languages earlier than 4-5 kya, meaning that the earliest settlers of the area have spoken another language. Linguistic evidence shows that Saami languages were spoken in the whole Finnish region until 1000 CE. According to historical sources, Lapps lived in central Finland in the 1500s but it is unknown if they were original Saami or Finns that have converted from agriculture to hunting and fishing.
Lamnidis et al. study samples from Levänluhta, a spring in Isokyrö, in the Finnish west coast, and an Iron Age water graveyard. The soil in Finland is naturally so acidic that ancient DNA doesn’t survive and the Levänluhta site is one of the few places from where genomes of ancient humans have been successfully studied. The Levänluhta samples are dated to be from 300-800 CE and thus expected to show affinity to modern-day Saami. That is indeed the case, and all but one of the individuals are close to modern Saami in the PCA; one of them wasn’t of local ancestry at all but groups with Western Europeans:
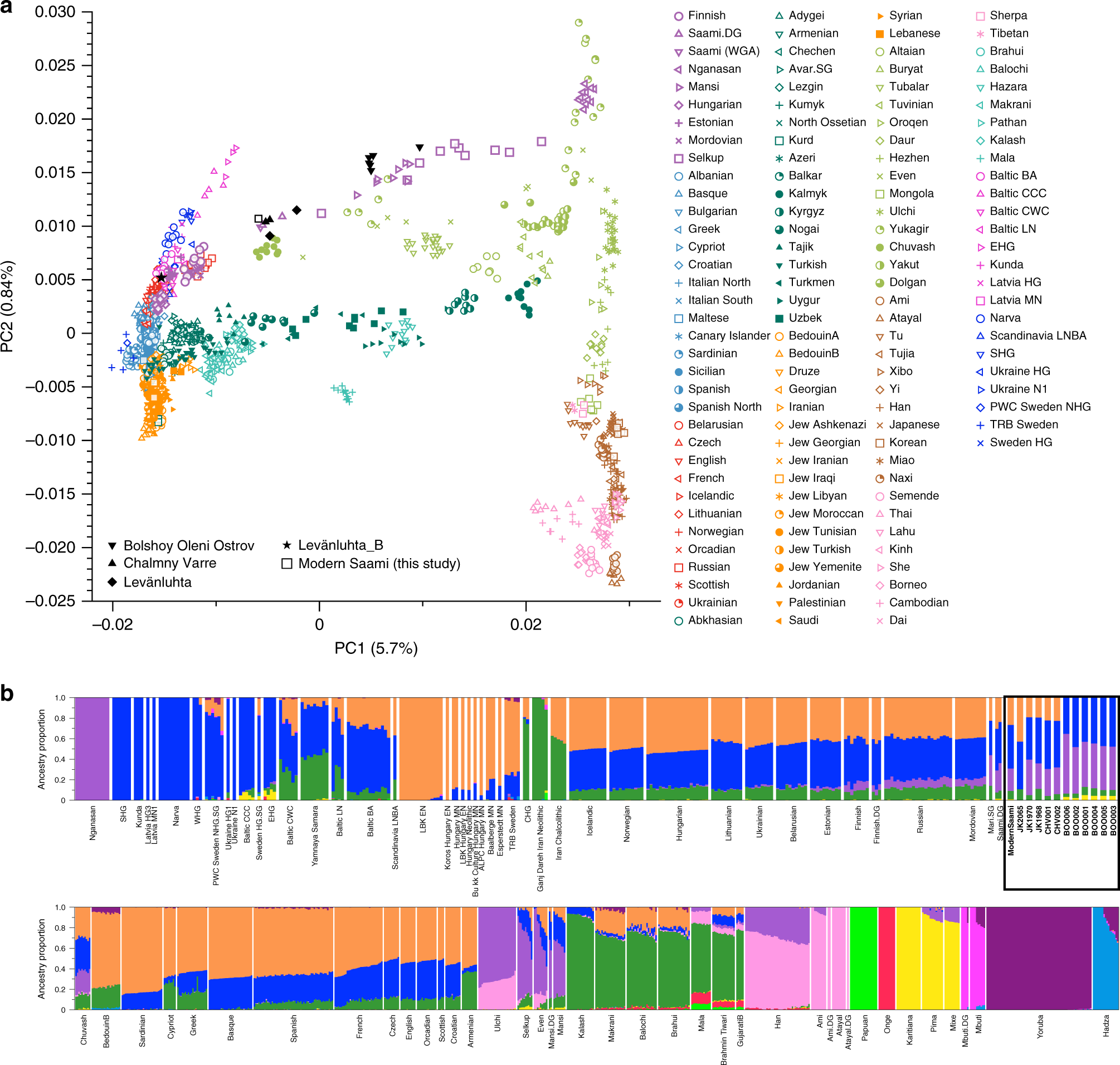
The ADMIXTURE plot allows studying ancestry components shared by other populations, especially those associated with the different ancestry sources of Europeans. However, that can be better tested using F-statistics with the program qpAdm. That shows that Finns have a greater proportion of their ancestry associated with Nganasans than the Central and Western European populations. That doesn’t of course mean direct migration between the populations but rather gene flow to Finns (and some other European populations) from a Siberian population ancestral to modern-day Nganasans.
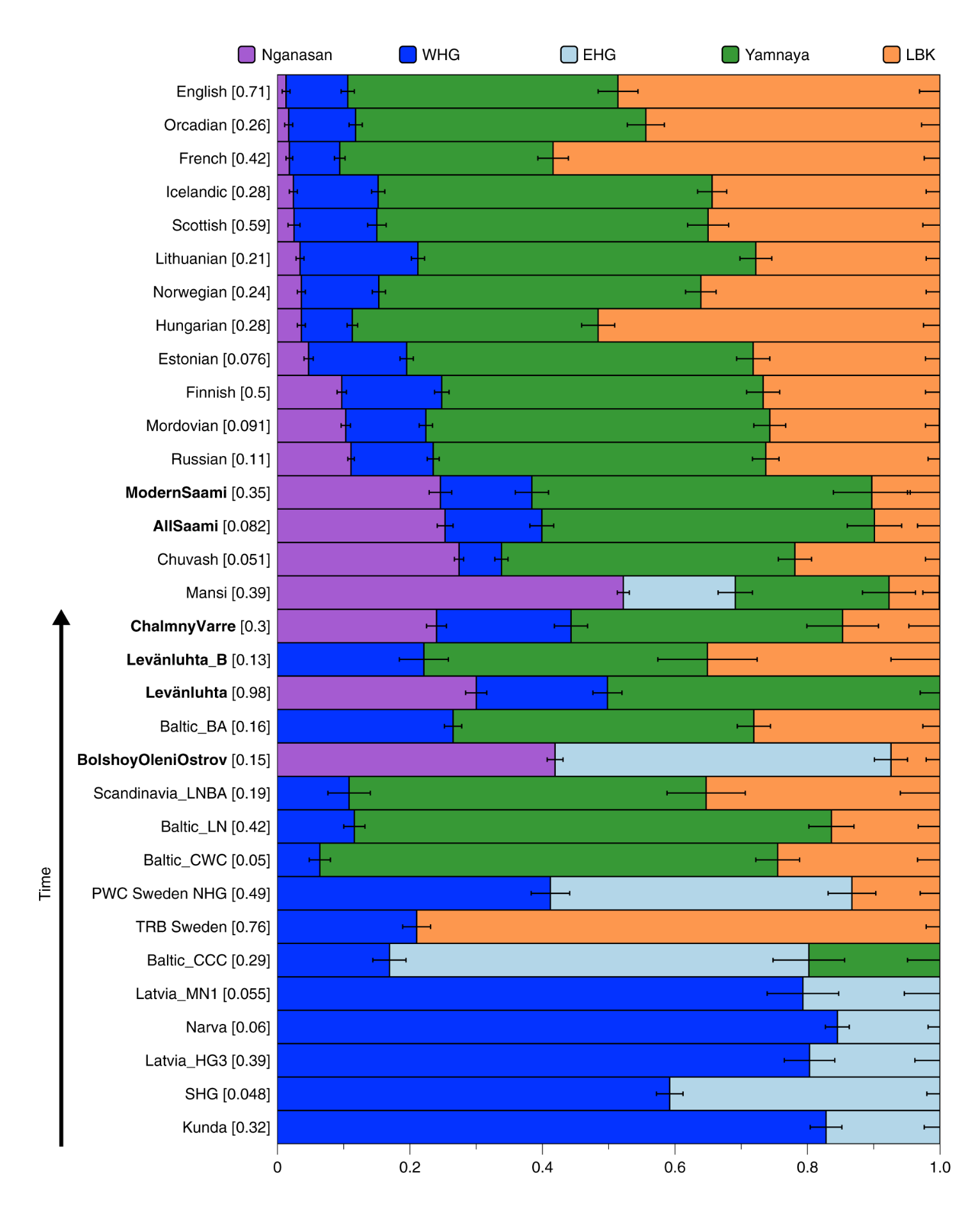
The Nganasan-associated ancestry is not present in ancient hunter-gatherers from the Baltics (8000 ya) and Sweden (~8000 ya). Using an archaic sample the Bolshoy Oleni Ostrov site, the ancestry was estimated to have been introduced 4000 ya. It has then spread to neighbouring populations, getting diluted on the way.
PCA is a multivariate analysis that reduces the complexity of datasets while preserving data covariance. It was introduced by Karl Pearson in 1901 and popularised in population genetics by Luca Cavalli-Sforza in the late 1970s. It was criticised at the time and claimed of not proving the human migrations patterns as Cavalli-Sforza wrote. The method was little used before Price et al. introduced smartPca and Novembre et al. claimed that genes mirror geography. It has then become a central tool in the analyses of very large population genetic data.
PCA is mathematically correct and is highly useful in quality control and outsider detection. However, PCA is shown to be extremely sensitive to sample set up and the results may dramatically change when the number of samples from different populations is altered. Crucially, the method can be adjusted to give the expected result: this can happen insincerely to get a desired outcome, or naively by researchers tuning the analysis until the result “makes sense”. As such, PCA should not be used alone to infer admixture, population of origin, or evolutionary histories.
Genetic information of thousands of humans – both modern and archaic – is freely available in public repositories. The analyses of these have enabled to construct increasingly complex and precise models of human history. Although Finns have been heavily investigated for medical purposes, DNA is preserved poorly in the local soil and the genomes of only few archaic samples have been resolved.