9. Human data at 1KGP and AADR
After this chapter, the students can retrieve 1000 Genomes Project data and Allen Ancient DNA Resource data and perform analyses on them.
Getting to Puhti
Get to your working directory:
$ WORK=/scratch/project_2000598
$ cd $WORK/$USERIf you are on a login node (if you access Puhti through a stand-alone terminal, you get to a login node; in the web interface, you can select either a login node or a compute node), start an interactive job (see sinteractive -h for the options):
$ # the command below is optional, see the text!
$ sinteractive -A project_2000598 -p small -c 2 -m 4000 -t 8:00:001000 Genomes Project data
The 1KGP data are available through the new URL at www.internationalgenome.org. There are fancy methods to browse the data but for own analyses one has to get the data through the FTP site at ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections. The site appears to provide also HGDP and SGDP data but those are only available in a raw (BAM) format. We select the latest 1KGP directory, 1000G_2504_high_coverage.
Make a new directory for the human analyses:
$ cd $WORK/$USER
$ mkdir humans
$ cd humansand download the metadata file:
$ wget https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/20130606_g1k_3202_samples_ped_population.txt -O 1kgp_meta.txt
$ less -S 1kgp_meta.txtThe actual data are under the directory working/. There are multiple versions of the data and, again, we select the latest with an appropriate name, 20220422_3202_phased_SNV_INDEL_SV. This seems to indicate that the data are phased and contain SNVs (SNPs), indels and structural variants. We are only interested in the binary indels and download them for the first 25 Mbp in chr1. Open the FTP directory in a browser, right-click the first file (with the name “1kGP….chr1…vcf.gz”) and select “Copy link address”. Paste the address in the command below.
$ module load samtools
$ URL=<paste the URL here>
$ bcftools view -v snps -m2 -M2 -r chr1:1-25000000 $URL -Oz -o 1kgp.chr1_snps.vcf.gzbcftools and samtools can read the data from a remote URL and this should now create a local with the chosen filtering applied. Note that the file will be 235 MB in size and downloading will take a while.
Once this is done, process the file with plink without and with a MAF filter:
$ module load plink/1.90b7.7
$ plink --vcf 1kgp.chr1_snps.vcf.gz --pca --freq gz --out 1kgp_chr1_25M
$ plink --vcf 1kgp.chr1_snps.vcf.gz --maf 0.05 --pca --freq gz --out 1kgp_chr1_25M_maf05Note that the VCF doesn’t include ancestral alleles and allele frequency analyses have to use folding; then the maximum MAF limit (not set here) must be below 0.5.
PCA
One completed, plot the PCA in R:
setwd(paste0("/scratch/project_2000598/",Sys.getenv("USER"),"/humans"))
library(ggplot2)
library(dplyr)
meta <- read.table("1kgp_meta.txt", header=T)
vec <- read.table("1kgp_chr1_25M.eigenvec",col.names = c("smp","pop",paste0("PC",1:20)))
vec$pop <- meta$Population[match(vec$smp,meta$SampleID)]
vec$spop <- meta$Superpopulation[match(vec$smp,meta$SampleID)]
val <- read.table("1kgp_chr1_25M_maf05.eigenval",header=F)[,1]
lab1 <- paste0("PC1 (",round(val[1]/sum(val)*100,2),"%)")
lab2 <- paste0("PC2 (",round(val[2]/sum(val)*100,2),"%)")
# colors for Population
ggplot(vec) +
geom_point(aes(PC1,PC2,color=pop)) +
xlab(lab1) + ylab(lab2) +
theme_classic()
# colors for Superpopulation
ggplot(vec) +
geom_point(aes(PC1,PC2,color=spop)) +
xlab(lab1) + ylab(lab2) +
theme_classic()The clear separation of the EAS superpopulation looks a bit surprising. If you test to plot it again after MAF>0.05 filtering, the separation is not quite as significant. Probably it caused by the fact that EAS is represented by two clean populations, CHB and JPT. SAS is represented by five populations and some of these were sampled in exile, thus potentially containing foreign ancestry.
Indeed, the impact of admixture can be seen among the AFR samples. Some of these are from Africa while some others are of African ancestry but now living in the Americas:
ggplot() +
geom_point(data=vec,aes(PC1,PC2),color="gray") +
geom_point(data=subset(vec,spop=="AFR"),aes(PC1,PC2,color=pop)) +
xlab(lab1) + ylab(lab2) +
theme_classic()The figure below shows the PCA with two populations highlighted. Compare that to your plot and answer the questions in Moodle.
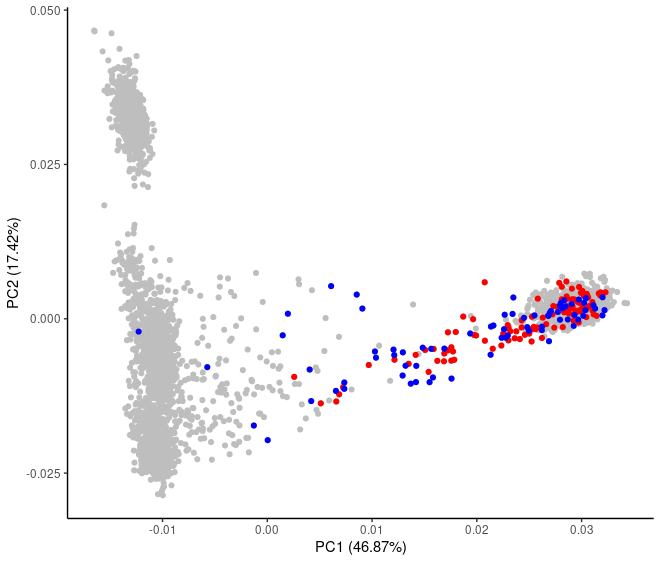
Exercise 1 in Moodle.
PCA is mathematically correct but shown to be extremely sensitive to sample set up and the results may dramatically change when the number of samples from different populations is altered. Because of that, PCA should not be used alone to infer admixture, population of origin, or evolutionary histories.
SAF
In the context of derived allele frequencies, it was mentioned that the SAF distributions should show the footprints of negative selection. We can test that with the 1KGP data.
In the FTP directory, there are other datasets and one of them ends with “_with_annot”. We download their annnotation of coding variants in chr1:
$ wget https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20201028_3202_raw_GT_with_annot/20201028_CCDG_14151_B01_GRM_WGS_2020-08-05_chr1.recalibrated_variants.annotated.coding.txt -O annotation.chr1.txtOur plink analysis computed the minor allele frequencies for variant positions. Strangely, this doesn’t have the typical CHROM and POS columns but the SNP name consisting of the chromosome, position and the alleles:
$ less -S 1kgp_chr1_25M.frq.gzNevertheless, we can extract the variants listed in the annotation file and write them into another file:
$ zgrep -f <(awk '{print gsub("chr","",$1)":"$2}' annotation.chr1.txt) 1kgp_chr1_25M.frq.gz > 1kgp_chr1_25M_CDS.frqWe can now plot the SAF for the full data and the CDS variants only:
frq.full <- read.table("1kgp_chr1_25M.frq.gz",header=T)
frq.cds <- read.table("1kgp_chr1_25M_CDS.frq",col.names = colnames(frq.full))
hist.full <- hist(frq.full$MAF,breaks=seq(0,0.5,0.01),plot=F)
hist.cds <- hist(frq.cds$MAF,breaks=seq(0,0.5,0.01),plot=F)
frq <- rbind.data.frame(
data.frame(set="full",mids=hist.full$mids,density=hist.full$density),
data.frame(set="cds",mids=hist.cds$mids,density=hist.cds$density)
)
ggplot(frq) +
geom_col(aes(x=mids,y=density,fill=set),position = "dodge") +
theme_classic() + xlim(0,0.15)As the data are from diverse populations, it contains a huge number of singletons. Despite that, the expected shift in the distributions is visible.
Allen Ancient DNA Resource data
The latest version of AADR is available at the shared data directory:
$ ls -lh $WORK/shared/humans/v62.0_1240k_public.*Due to its size, we only make links to the full data:
$ cd $WORK/$USER/humans
$ mkdir aadr
$ cd aadr
$ ln -s $WORK/shared/humans/v62.0_1240k_public.* .The files “*.ind”, “*.snp” and “*.geno” sound familiar and are similar to those generated in earlier practicals from VCF data. The file “*.anno” contains extended annotation of the individuals in the data. We can see the columns:
$ head -1 v62.0_1240k_public.anno | sed 's/\t/\n/g' | grep -n .We could select the IDs, age, locality, coordinates and SNP counts and print them out:
$ tail -n+2 v62.0_1240k_public.anno | cut -f1-2,10,15-18,23,24 | head | column -s$'\t' -t | less -SPCA
To make the files smaller and analyses faster, we reduce the data and keep only chromosome 1:
$ module load eigensoft
$ cat > convert.txt << EOF
genotypename: v62.0_1240k_public.geno
snpname: v62.0_1240k_public.snp
indivname: v62.0_1240k_public.ind
chrom: 1
genotypeoutname: global_chr1.geno
snpoutname: global_chr1.snp
indivoutname: global_chr1.ind
EOF
$ convertf -p convert.txt This may take up to 10 minutes.
The “*.anno” file contains crucial data but is really messy. We can collect the most important columns into a separate file:
$ cut -f1-2,10,16-18 v62.0_1240k_public.anno > global.metaIn the analyses of ancient DNA, some samples may miss significant proportions of variant positions. The fact that samples are missing lots of information may make them to appear more similar in analyses such as PCA. It is therefore recommended that the PCs are first estimated with good quality data and then the possibly problematic samples are projected on those.
The AADR website says this about the sequencing quality:
SG=samples with whole genome shotgun sequence data, randomly drawing a single read to represent each position in the genome
DG=samples shotgun sequenced with high enough coverage to call diploid genotypes, allowing for heterozygous callsWe can see that around ~20% of the samples are “DG” and modern; the column 41 tells whether the quality is OK:
$ awk -F"\t" '$1~/DG/ && $10==0 && $41=="PASS"' v62.0_1240k_public.anno | wc -l
$ awk -F"\t" '$1~/DG/ && $10==0 && $41=="PASS" {print $16}' v62.0_1240k_public.anno | sort | uniq -cHowever, the number of samples per locality varies greatly and partly that is explained by the 1KGP data:
$ awk -F"\t" '$1~/DG/ && $10==0 && $14~/[A-Z]{3}.DG/ && $41=="PASS" {print $14,$16}' v62.0_1240k_public.anno | sort | uniq -cThey may be fine data and worth retaining in more serious analyses but here we remove them from our initial PCA data. We generate a list of populations to retain in the computation of PCA as:
$ awk -F"\t" '$1~/DG/ && $10==0 && $41=="PASS"{print $14}' v62.0_1240k_public.anno | sort | grep -v -e Ignore -e [A-Z][A-Z][A-Z].*.DG | uniq > global_DG.pops.txtThen, we can create the control file for smartpca and execute it:
$ cat > smartpca.DG.txt << EOF
genotypename: global_chr1.geno
snpname: global_chr1.snp
indivname: global_chr1.ind
evecoutname: global_chr1.DG.evec.txt
evaloutname: global_chr1.DG.eval.txt
poplistname: global_DG.pops.txt
lsqproject: YES
numoutevec: 4
EOF
$ smartpca -p smartpca.DG.txt > smartpca.DG.outOnce that is finished, we can visualise the results in R:
setwd(paste0("/scratch/project_2000598/",Sys.getenv("USER"),"/humans/aadr"))
library(ggplot2)
library(dplyr)
vec <- read.table("global_chr1.DG.evec.txt",col.names=c("smp",paste0("PC",1:4),"pop"))
meta <- read.table("global.meta",sep="\t",col.names=c("ID","Name","Age","Country","Lat","Long"))
k <- match(vec$smp,meta$ID)
vec <- cbind.data.frame(vec,meta[k,])
# select only the DG samples for which coordinates are given
vec.dg <- vec %>% filter(grepl("DG",ID),Age==0,Lat!="..",Long!="..")
vec.dg$Lat <- as.numeric(vec.dg$Lat)
vec.dg$Long <- as.numeric(vec.dg$Long)
# and create a function that defines a colour based on them
range01 <- function(x){(x-min(x))/(max(x)-min(x))}
make_rgb <- function(x,y){
rgb(range01(x),range01(y),1)
}
val <- read.table("global_chr1.DG.eval.txt")[,1]
lab1 <- paste0("PC1 (",round(val[1]/sum(val)*100,2),"%)")
lab2 <- paste0("PC2 (",round(val[2]/sum(val)*100,2),"%)")
# plot the samples with colours from coordinates
ggplot(vec.dg) +
geom_point(aes(PC1,PC2),fill=make_rgb(vec.dg$Lat,vec.dg$Long),shape=21,alpha=0.5) +
xlab(lab1) + ylab(lab2) + theme_classic() + guides(fill="none")
# and again with some populations highlighted
ggplot(vec.dg) +
geom_point(aes(PC1,PC2),fill=make_rgb(vec.dg$Lat,vec.dg$Long),shape=21,alpha=0.5) +
geom_point(data=subset(vec.dg,Country=="Italy"),aes(PC1,PC2),fill="magenta",shape=21) +
geom_point(data=subset(vec.dg,Country=="Finland"),aes(PC1,PC2),fill="white",shape=21) +
geom_point(data=subset(vec.dg,Country=="Japan"),aes(PC1,PC2),fill="red",shape=21) +
geom_point(data=subset(vec.dg,Country=="Nigeria"),aes(PC1,PC2),fill="green",shape=21) +
xlab(lab1) + ylab(lab2) + theme_classic() + guides(fill="none")The PCA for the full data doesn’t look much different except that the distribution is more continuous:
# select again samples with coordinate information
vec.all <- vec %>% filter(Lat!="..",Long!="..",!is.na(Lat),!is.na(Long))
vec.all$Lat <- as.numeric(vec.all$Lat)
vec.all$Long <- as.numeric(vec.all$Long)
# plot them with colours from coordinates
ggplot(vec.all) +
geom_point(aes(PC1,PC2),fill=make_rgb(vec.all$Lat,vec.all$Long),shape=21,alpha=0.5) +
xlab(lab1) + ylab(lab2) + theme_classic() + guides(fill="none")
# and again with some highlighted populations
ggplot(vec.all) +
geom_point(aes(PC1,PC2),fill=make_rgb(vec.all$Lat,vec.all$Long),shape=21,alpha=0.5) +
geom_point(data=subset(vec,Country=="Italy"),aes(PC1,PC2),fill="magenta",shape=21) +
geom_point(data=subset(vec,Country=="Finland"),aes(PC1,PC2),fill="white",shape=21) +
geom_point(data=subset(vec,Country=="Japan"),aes(PC1,PC2),fill="red",shape=21) +
geom_point(data=subset(vec,Country=="Nigeria"),aes(PC1,PC2),fill="green",shape=21) +
xlab(lab1) + ylab(lab2) + theme_classic() + guides(fill="none")The position of Finnish samples looks surprisingly spread out. A closer look on the samples reveals that all samples from Finland are not “Finns”:
vec %>% filter(Country=="Finland") %>% select(pop) %>% unique()The figure below shows the PCA for the vec.dg data. Compare that to your plot and answer the questions in Moodle.
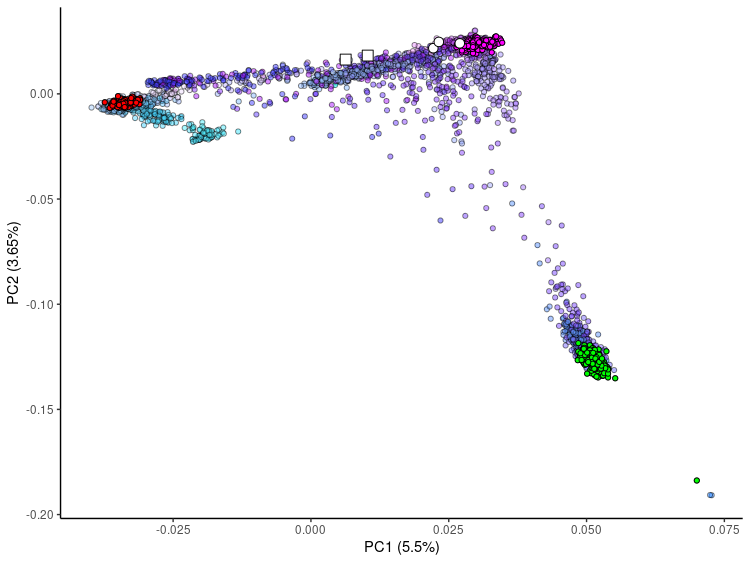
Exercise 3 in Moodle.
PCA is mathematically correct but shown to be extremely sensitive to sample set up and the results may dramatically change when the number of samples from different populations is altered. Because of that, PCA should not be used alone to infer admixture, population of origin, or evolutionary histories.
Frequency change over time
The SNP “rs4988235” is associated with the ability to digest milk as an adult. As a result of positive selection, it is common in many parts of Europe. Ideally, we should see the beneficial allele increasing in frequency in the AADR data.
The SNP is in the data with its ID:
$ grep rs4988235 v62.0_1240k_public.snp The SNP ID cannot be directly used to extract data but its genomic coordinates can be set as the limits; it’s also useful to output the genotypes in a human readable Eigenstrat format:
$ cat > convert.txt << EOF
genotypename: v62.0_1240k_public.geno
snpname: v62.0_1240k_public.snp
indivname: v62.0_1240k_public.ind
outputformat: EIGENSTRAT
chrom: 2
lopos: 136608646
hipos: 136608646
genotypeoutname: rs4988235.geno
snpoutname: rs4988235.snp
indivoutname: rs4988235.ind
EOF
$ convertf -p convert.txt The “*.anno” file is messy and it’s sufficient to extract just a few columns:
$ cut -f1-2,10,14,16-18,41 v62.0_1240k_public.anno > rs4988235.metaWith those, we can start analysing the data in R:
setwd(paste0("/scratch/project_2000598/",Sys.getenv("USER"),"/humans/aadr"))
library(tidyr)
library(dplyr)
library(ggplot2)
# read genotypes, individuals and meta data
gt <- do.call(rbind,lapply(strsplit(scan('rs4988235.geno',what=""), ""), as.numeric))
ind <- read.table('rs4988235.ind',header=F)
colnames(ind) <- c('id','sex','pop')
meta <- read.table('rs4988235.meta',sep='\t',skip=1, quote='')
colnames(meta) <- c("GeneticID","MasterID","Age","GroupID","PoliticalEntity","Lat","Long","Quality")
meta$Long <- as.numeric(meta$Long)
meta$Lat <- as.numeric(meta$Lat)In these analyses, we focus on European populations only. Instead of listing the names of countries (a viable option, though), we select the samples based on their coordinates and require their quality to be “PASS”. The age of the samples varies greatly and we bin them to rough time periods:
meta.europe <- meta %>% filter(Lat > 35, Lat < 72, Long > -10, Long < 61, Quality=="PASS", GeneticID %in% ind$id)
keep <- ind$id %in% meta.europe$GeneticID
meta.europe$GT <- gt[1,keep]
meta.europe <- meta.europe %>% mutate(Period = cut(Age,breaks=c(-1,500,1500,2500,3500,5000,10000),labels = c(0,500,1500,2500,3500,5000)))Next we check which countries have enough samples across the different time periods. The first command gives a list Country-Period combinations with at least five samples. From these, we manually select a number of potentially interesting countries (vector “ctr”) and then process the data:
meta.europe %>% select(PoliticalEntity,Period) %>% unite(group,sep="-") %>% table() %>% as.data.frame %>% filter(Freq>5)
ctr <- c("Armenia","Croatia","Czechia","Denmark","Estonia","France","Germany","Hungary","Italy","Poland","Russia","Spain","Sweden","United Kingdom")
meta.europe %>% filter(PoliticalEntity %in% ctr) %>% select(PoliticalEntity,Period,GT) %>%
group_by(PoliticalEntity,Period) %>% reframe(PoliticalEntity,Period,AF=mean(GT)/2) %>%
distinct() %>% spread(Period,AF)The last command first filters the data and keeps only the samples from focal countries, selects the columns of interest, groups the samples by country & period and then computes a new variable “AF” as the mean of GTs; of the results, only one line of each “country & period” is retained and that is converted to the wide format.
When reading the resulting table, note that the allele frequency is for the ancestral allele. The allele giving ability to digest lactose is the derived allele. It indeed increases in frequency toward the current day but only in some parts of Europe.
Finally, we can plot the allele frequencies at periods “2500” and “500” on a map. The code for that is a bit messy and not explained in detail:
library(rnaturalearth)
library(sf)
library(viridis)
world <- ne_countries(scale = "medium", returnclass = "sf")
d.500 <-
meta.europe %>% filter(PoliticalEntity %in% ctr & Period==500) %>% select(PoliticalEntity,Period,Lat,Long,GT) %>%
group_by(PoliticalEntity,Period) %>% reframe(PoliticalEntity,Period,Lat=mean(Lat),Long=mean(Long),AF=mean(GT)/2) %>%
distinct(PoliticalEntity,Period,.keep_all = TRUE) %>% mutate(AF=1-AF)
d.2500 <-
meta.europe %>% filter(PoliticalEntity %in% ctr & Period==2500) %>% select(PoliticalEntity,Period,Lat,Long,GT) %>%
group_by(PoliticalEntity,Period) %>% reframe(PoliticalEntity,Period,Lat=mean(Lat),Long=mean(Long),AF=mean(GT)/2) %>%
distinct(PoliticalEntity,Period,.keep_all = TRUE) %>% mutate(AF=1-AF)
dat <- left_join(d.500,d.2500,join_by(PoliticalEntity)) %>% st_as_sf(coords = c('Long.x','Lat.x')) %>%
st_set_crs(4133) %>% rename(AF.500=AF.x,AF.2500=AF.y) %>% select(PoliticalEntity,AF.500,AF.2500,geometry)
ggplot(data = world) +
geom_sf(fill= "gray90",col=gray(0.65),size=0.05) +
geom_sf(data = dat, aes(fill=AF.2500),size=8,shape=21)+
geom_sf(data = dat, aes(fill=AF.500),size=4,shape=21)+
coord_sf(crs="+init=epsg:3035 +proj=laea +lat_0=60 +lon_0=27 +x_0=4321000 +y_0=3210000 +ellps=GRS80 +towgs84=0,0,0,0,0,0,0 +units=m +no_defs",
xlim = c(1000000, 5000000), ylim = c(500000, 5000000), expand = FALSE)+
theme(
panel.background = element_rect(fill = "aliceblue"),
panel.grid = element_line(size=0.05,color = gray(.35),linetype = 'dashed')
) +
scale_fill_gradientn(colours = rev(viridis(20)),limits=c(0,1)) + labs(fill="")Note that now the AF values have been reversed.
There are several open resources for human genetic data. www.internationalgenome.org, formerly “1000genomes.ebi.ac.uk”, provides raw data and several versions of VCF files for a few thousand humans from a few tens of populations. The Allen Ancient DNA Resource provides the genotype data analysed in numerous recent human population genetic studies. No raw data and few details of data processing are provided. The current version of AADR “1240k” data contain ~1.23M variant positions for ~17,600 individuals; AADR “HO” data contain ~584k variant positions for ~22,000 individuals. Many individuals are provided as pseudo-haploids; the data set contains lots of missing data.