Why population genomics?
What is “population genetics”?
Population genetics studies changes in allele frequencies within and between populations. Population can be any renewing group of organisms. Factors influencing the allele frequencies – and thus studied by population genetics – include:
- genetic drift
- gene flow: migration, admixture, introgression
- positive and negative selection
- dominance vs. recessiveness
- linkage and epistasis
- mutations
Population genetics can look back in time and infer the mechanisms that created the observed patterns, measure the changes happening now, or predict what will happen in the future.
Foundations and early history
Population genetics as a discipline is based on the findings of Charles Darwin and Gregor Mendel. Initially, Darwin’s natural selection and Mendel’s laws of inheritance were thought to be incompatible with each other: the first was based on minute steps by selection while the latter explained how existing – often wildly different – features were transferred from parents to the offspring. The work in the early 20th century, called the Great Synthesis, showed that with an interplay of multiple genes, Mendel’s inheritance can explain the Darwinian gradual change. The central figures in this work were Ronald Fisher, Sewall Wright, Thomas Hunt Morgan and John Haldane.
Population genetics is the closest that any biological field gets to theoretical physics: it has a solid theoretical foundation that allows devising experiments as well as evaluate observations. For a long time, population genetics was a rich collection of theories that could not properly be tested in practice as measuring the allele frequencies in large scale wasn’t feasible. This has now changed with the development of high-throughput genome sequencing and, probably first time ever, the amount of data surpasses researchers’ ability to analyse them.
Ronald Fisher and Sewall Wright, the fathers of population genetics, did their work in the first half of 20th century. The second half of the century included significant findings, such as the neutral theory by Motoo Kimura, but the first truly revolutionary novel concept was the coalescent theory, developed independently in 1980s by John Kingman, Richard Hudson and Fumio Tajima. Importantly, the coalescent theory was especially well suited for the analysis of the modern single-nucleotide polymorphism (SNP) data produced by the high-throughput genome sequencing.
Alleles and their frequencies
If population genetics studies allele frequencies, what is an allele? The term allele is an abbreviation of allelemorph and was coined in the early 20th century by two British geneticists, William Bateson and Edith Saunders, coming originally from Greek words allelo- or allos, meaning alternative or other. With this term, they referred to the alternative “forms” described by Mendel, such as “pea shape” being either “round” or “wrinkled”. Mendel understood that these forms are caused by some genetic factors but didn’t have more precise names for these “factors”. We now know that the differing forms are caused by alternative alleles of a gene.
The field of genetics can be divided into Mendelian genetics studying the mechanisms of inheritance and molecular genetics studying the molecules and molecular processes behind that. Due to that division, some central terms have double meaning; gene means both the inherited entity producing a certain feature and a sequence of DNA; similarly, allele can mean both the phenotypic output, such round or wrinkled peas, or the alternative molecular forms producing that output. In the age of genomes, an allele typically means the alternative nucleotides at a specific position of a genome.
Alleles in the genomic era
As an example, the variant at position 89920138 in the human chromosome 16 is known as “rs1805009”. The ancestral allele at this position is the nucleotide G; in some European and American populations an alternative allele, nucleotide C, is present at very low frequencies. The base change from G to C causes an amino acid change from aspartate to histidine at position 294 of the protein produced by the MC1R gene (Melanocortin 1 receptor). This one base change produces red hair in people carrying the C allele. In other words, C and G are alleles; the alternative forms of the MC1R gene can be considered alleles; and the red hair colour can be considered the “red hair” allele.

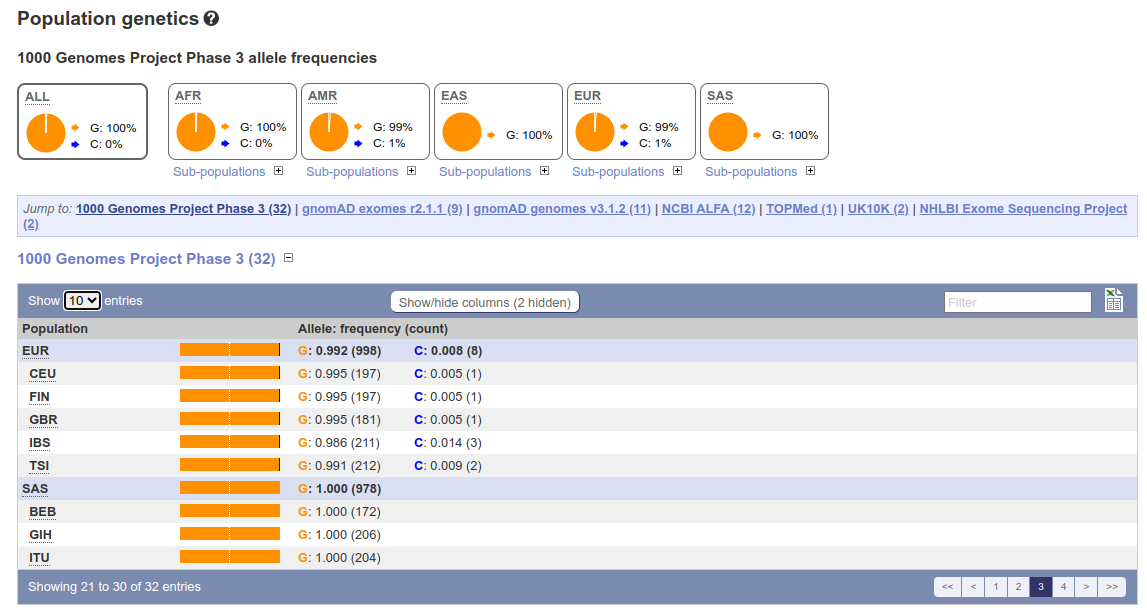
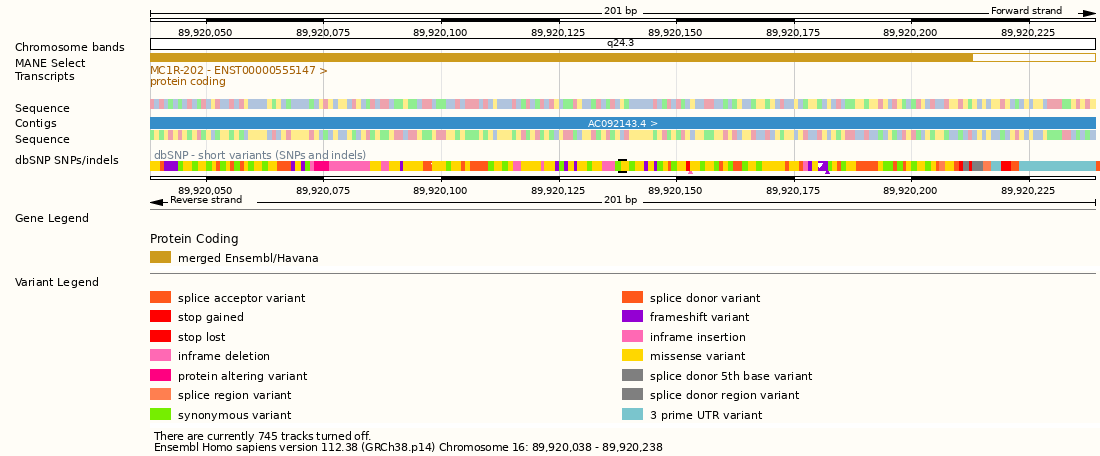
On this course, we focus on studying the narrowest definition, such as the frequencies of G and C at position 16:89920138. We will learn how to find the allele combinations, i.e. the genotypes, of all genomic loci of an individual, how to compute summary statistics of allele frequencies in populations, and how to perform population genetic analyses based on allelic data and information. We do the analyses using large data, either from full genomes or significant parts of the genome. Given the genomic scale, the course is called Population Genomic Data Analysis.
The two dimensions of population genetic data
The population genetic data handled on this course are genomic sequences or, more precisely, variants within genomic sequences segregating among the study samples. There are two dimensions on this kind of data: across the genomic sites and across the study samples.
Mutations are a random process that one could think as snow flakes falling on top of (1) the sequences or (2) the tree relating the sequences.
In the figure below, the magenta “flakes” are falling horizontally, and the left one is hitting a “gene” while the right one falls through and affects the sequences below. The left mutation did happen but, as it changed the gene and the resulting protein, it was lethal: the individual carrying the mutation died before producing offspring and we cannot observe it in our data. By accumulating lots of data, One can utilise the mutation patterns to learn about the structural constraints of the genome. By assuming the mutation process fairly random across the genome (this is not entirely true), we can infer the impact of natural selection on different genomic regions and features. With lots of data, one could infer the locations of unknown genomic features based on deviations in the mutation patterns.

On the left side, we should imagine the tree laying flat on a table and some of the flakes – falling from above – randomly hitting the tree edges. There are two important points to notice from this: (i) the chances of a mutation hitting a particular tree edge (black lines) depends on the edge length: the longer the edge, the more likely the flake falls on it; and (ii) the mutation is inherited by all the descendants of that particular edge (here, three sequences). However, there’s a major caveat on this.
Due to recombination, there is no single tree that relates the sequences in a correct manner. In principle, every single genomic site could have a tree of its own; in practice, blocks of unknown length share the same tree and the tree for the neighbouring block is is similar but not identical. In population genetics, the tree is a hidden parameter that we know exists (all sexual individuals are related by a tree representing their ancestry) but that we rarely can infer. Even if the sequences are identical, they must be related by a tree.
We’ll come back to these two dimensions of the data on the course.
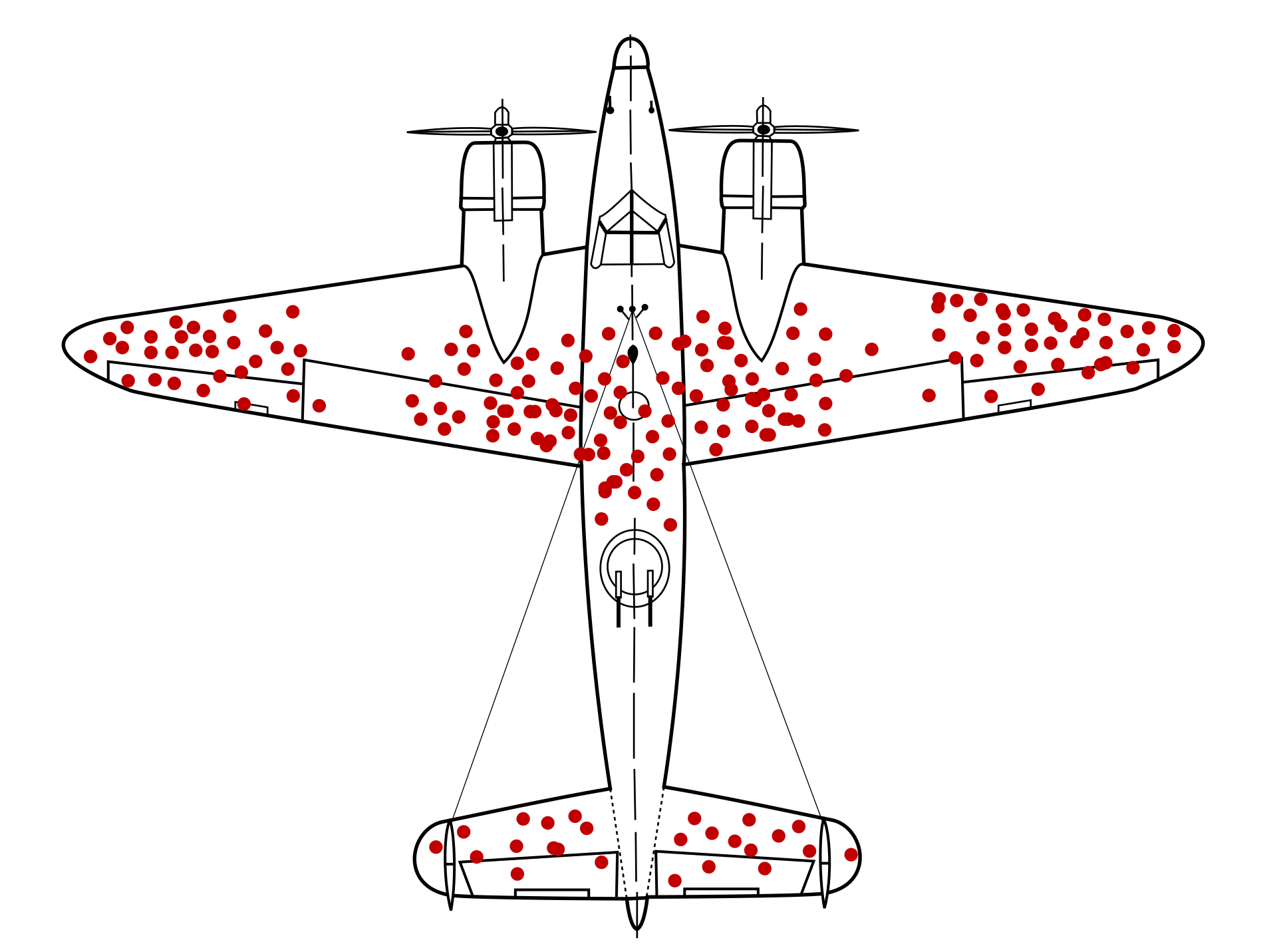
The first dimension – across the sites of a genome – can be considered information about “survivorship”. In a famous case, the allied airforces suffered lots of casualties in WW2 bomber missions. To improve their planes, they studied the damage in the returning planes and added protection to areas that were hit the most. That made the planes heavier but didn’t improve their chances of returning from missions. It was the insight from statisticians that one should rather add armour to areas that show the least damage – these are the most important parts of the plane.
All of the data handled on this course is from living individuals and, similarly to planes, the observed mutations cannot have been lethal.
Why population genomics matters?
There are many good reasons to do this course:
Basically all biological disciplines are going through an explosion in data quantities and are becoming computational. “Genomics” are the forefront of this and are producing huge amounts of DNA sequencing or other measurement data. Learning computational data analysis with population genomic data is useful for many other fields of biology.
Population genetics is the foundation of and the connecting link between ecology and evolutionary biology. Modern population genetics is based on the analyses of genomic data.
In molecular biology and genetics, it is increasingly understood that studying single individuals can be misleading and it is important to understand the population level variation. When studying multiple individuals, it is important to understand the relationships between the individuals and the mechanisms that have formed them as the members of connected populations.
For students with mathematical or data analysis background, population genetics and genomics provide one of the best formalised biological disciplines for theoretical and computational research and an easy entry point to biological sciences.
Population genomics is a hot topic and touches us, humans, as a species. The modern DNA sequencing and analysis methods have enabled studying the history humans in an unprecedented detail. Although most of these revolutionary analysis methods are applied to human first, research on other organisms gains from this rapid development.
University of Helsinki has many active research projects in population genomics and need students with skills in computational analysis methods.
This is a cool course and worth the effort!