1. Reference genomes
After this chapter, the students can list properties of a reference genome that make the mapping of short reads to it more difficult.
Assembly of a reference genome and read mapping to an existing reference genome are two very different tasks. Moreover, the reference genome needs to be done only once, making the job suitable to specialised experts. On this course, we utilise exiting reference genomes. However, we briefly discuss the properties of genomes that may affect the downstream analyses utilising it.
Properties of genomes
The genomes of organisms have properties that can significantly affect our attempt to analyse them. Two obvious properties are the size of the genome and its repeat content.
Genome size
The genome sizes vary massively and, although the size of the genome correlates with the organisms’ complexity, the variation within life forms is huge. Organisms may have undergone genome duplications but, within classes of organisms, the extreme size ranges are mostly caused by repetitive elements. On this course, we will analyse genomic data from stickleback fish, pinnipeds and human. Human’s 3 Gbp and ringed seal’s 2.3 Gbp are pretty typical genome sizes for mammals while stickleback’s 440 Mbp is towards the smaller end for bony fishes.
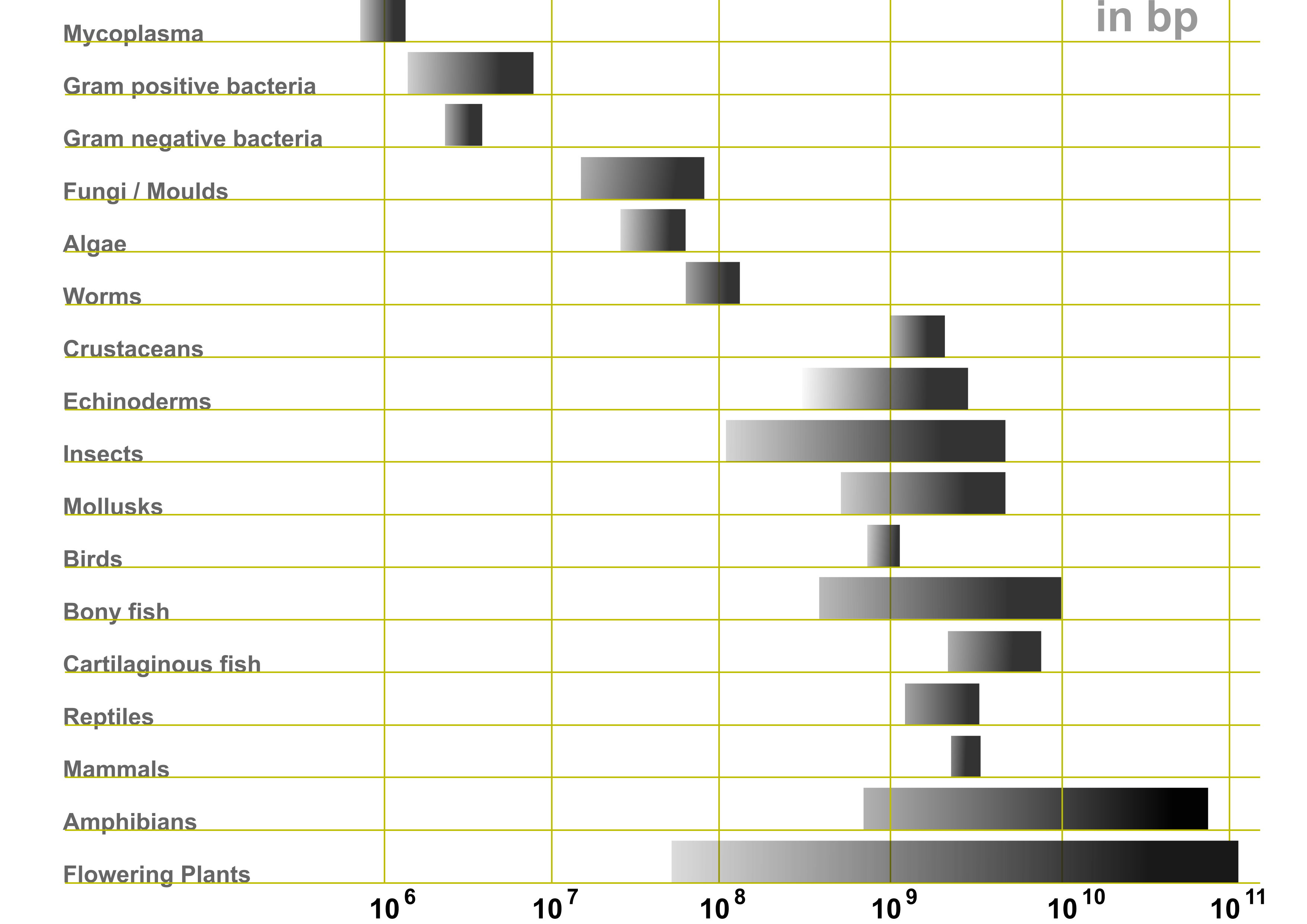
Large size of a genome increases the cost (DNA sequencing and computation) and the complexity of analyses, but a very small genome may not be ideal either. The reason is that many population genetic analyses assume the sites to evolve (nearly) neutrally and thus reflect processes such as gene flow or demographic history, not purifying selection and the function of the underlying sequence. For such analyses, we need non-functional and presumably neutrally evolving genome sites. Often we can’t know the functional importance of specific sites and just assume that the great majority of e.g. non-genic (=intergenic or intronic) sites evolve neutrally and the small percentage not doing so won’t affect our analyses. With very small genomes, this assumption may be incorrect.
Repeat types and content
Repeats can be divided into interspersed and non-interspersed. Interspersed repeats are caused by transposons creating copies of themselves and these copies integrating into new locations in the genome. Non-interspersed repeats are mainly short sequences that are repeated multiple times in a row, known as tandem repeats.
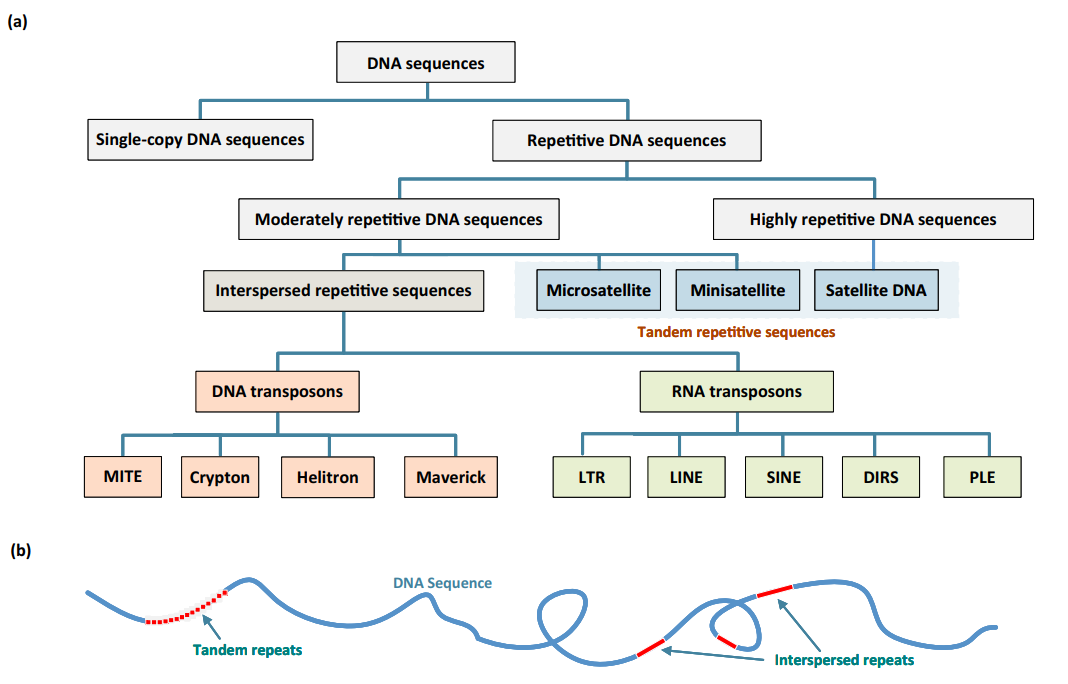
Repeat sequences can make a majority of a genome. In human, ringed seal and nine-spined stickleback, they make 40-50% of the current reference genomes (Table 1).
Table 1. Proportion of the human genome (Liao et al. 2023) and the nine-spined stickleback genome (Wang et al. 2024) consisting of non-repeat (single-copy) and repeat sequences of different type .
| Sequence type | Human | Ringed seal | Stickleback |
|---|---|---|---|
| Single-copy DNA | ~50 % | ~56 % | ~61 % |
| DNA transposons | ~5 % | ~3 % | ~14 % |
| RNA transposons LTRs | ~9 % | ~5 % | ~5 % |
| RNA transposons SINEs | ~13 % | ~9 % | ~1 % |
| RNA transposons LINEs | ~21 % | ~23 % | ~7 % |
| Tandem repeats | ~3 % | ~2 % | ~7 % |
The repeat elements vary so much that it seems strange to even merge them under a single term. The simplest repeats are clusters of a single base such poly-A’s consisting of tens or hundreds of adenosines in a row. Slightly more complex repeats are microsatellites and other satellite sequences (Table 2). Some of these are functional such as the repeats forming the centromeres or the telomeres. One mechanism retaining tandemly-duplicated sequences is thought to be polymerase slippage that may skip over copies or template new copies of the existing ones, thus keeping the repeats identical.
Transposable elements are completely different and can be nearly considered as living entities. They may contain genes required for their copying and translocation while some are related to viruses. The sizes of transposons vary from ~300 bp (Alu) to ~10 kbp. The significance of this is clarified below.
Genome assembly
The first place where the repeats complicate genomic analyses is the assembly of the reference genome. Typically, the reference genomes aim to be haploid representations of the genome sequence such that each chromosome is present once in a maximally accurate form. Extremely few reference genomes achieve that and most are approximations of varying quality of the true content and order of the sequences. Repeat sequences are problematic due to their high similarity: they are like the mono-colour regions in a jigsaw puzzle, all the black or blue pieces looking alike. Assembling the unique regions of a genome (or a puzzle) is fairly straightforward but the presence of repeats breaks the assembly into many small fragments, known as contigs.
The development of DNA sequencing technology has massively increased the length of continuous DNA reads and rge quality of the reference genomes. The early genomes were done using short-read sequencing (especially Illumina) that resolved 100-150 bp from both ends of DNA fragments. Using fragment libraries of various sizes, one could produce read-pairs that belong very close to each other in the genome – allowing to resolve local sequence – or further apart – possibly allowing to combine near-by fragments separated by repeats. Modern DNA sequencing technologies can produce nearly perfect DNA reads of several tens of kilobases (PacBio HiFi) or somewhat noisy reads of even hundreds of kilobases (Oxford Nanopore).


With read sizes of e.g. 20 kbp, one could “read through” all transposable elements (typically varying between 0.4 ad 10 kbp) and thus know the unique sequence at both sides of the repeat. With that, one should be able to identify each transposon as unique and place it in the right location in the genome. However, not all flanking sequences are unique and some arrays of tandem repeats are much longer than the longest sequencing reads (Table 2). With the current technology, one cannot resolve the length of such clusters or distinguish the flanking regions of two similar clusters.
Table 2. Classes and length distribution of tandem repeats in the human genome (Liao et al. 2023). Note the lengths of the repeat arrays (last column).
| Class of TRs | Length of TR unit | Length of TR array |
|---|---|---|
| Telomeres | ~6 bp | ~10–15 kb |
| Tandem paralogous | ||
| rDNA | ~43 kb | ~3–6 Mb |
| Segmental duplications | ~1–400 kb | ~1kb–5Mb |
| Microsatellites | ~2–6 bp | ~10–100bp |
| Minisatellites | ~10–100bp | ~100bp–20kb |
| Satellites | ||
| Alpha satellite | ~171bp | ~0.2–8Mb |
| Beta satellite | ~68 bp | ~60–80kb |
| Gamma satellite | ~48–220bp | ~11–121kb |
| Satellite I | ~17–25bp | ~2.5kb |
| Satellite II | ~23–200bp | ~11–70kb |
| Satellite III | ~5bp | ~3.6kb |
| Satellite IV | ~35bp | ~25–530kb |
| Macrosatellites | ~100bp–5kb | ~300kb |
| Megasatellites | ~1–5kb | ~400kb |
In addition to classical repeats, also genes and genome regions can be considered repeats. The most obvious of these are ribosomal DNA (rDNA) that are a class of tandem repeats. In human, rDNAs are 45-kbp near-identical repeats placed n five clusters in chromosomes 13, 14, 15, 21 and 22. The number of rDNA copies varies among individuals and a typical diploid human genome has an average of 315 rDNA copies, with a standard deviation of 104 copies. The first near-complete assembly for the human rDNA clusters was obtained only very recently (Nurk et al. 2022) and in most other genomes the clusters are collapsed.
Genomes contain random segmental duplications, near identical copies of certain genome regions that possibly contain genes, and the presence and absence of these duplications may vary across individuals creating potentially phenotypic diversity. As we see later, near-identical segmental duplications are the most problematic in population genetics analyses.
Genomes are huge in size and typically contain abundant data for population genetic inferences. The mapping of short sequencing reads is prone to error in regions containing repetitive elements and other non-unique sequence. If the aim is to infer evolutionary processes affecting the samples and lineages, not those differently affecting the functional categories of genome sites (see here), it is easiest to simply discard the problematic genome regions from the analyses.