6. Reassembly, recalling and variant annotation
After this chapter, the students can extract reads of a specific region and reassemble them into contig(s). They can apply alternative approaches to transfer gene annotation information from a closely-related species and annotate variants based on that.
Getting to Puhti
Get to your working directory:
$ WORK=/scratch/project_2000598
$ cd $WORK/$USERIf you are on a login node (if you access Puhti through a stand-alone terminal, you get to a login node; in the web interface, you can select either a login node or a compute node), start an interactive job (see sinteractive -h for the options):
$ # the command below is optional, see the text!
$ sinteractive -A project_2000598 -p small -c 2 -m 4000 -t 8:00:00Depending on the utilisation of the Puhti cluster, the last step may take time. If it doesn’t progress in a reasonable time (a couple of minutes), you may skip it and work on the login node. The work below is computationally light and should not put much strain on the login nodes.
We will utilise the SLURM job scheduling system at Puhti. You can rehearse the vocabulary and concepts used at https://intscicom.biocenter.helsinki.fi/section5.html#running-jobs-on-a-linux-cluster
Reassembly
If we look at the gene annotations for our references genome, we see that one of the contigs contains mitochondrial genes:
$ zgrep "Name=mt-" reference/ninespine_annotation_sorted.gff3.gzTypical vertebrate mitochondrion should be \(\sim\) 16 kbp long and this contig is clearly not of the right length. It turns out that this contig has been mis-assembled.
We can quite easily re-assemble parts of the genome using the reads currently mapped on that region in the BAM data. We just need to extract the reads and run them through an assembler program. (Remember that BAM lines contain everything that is in FASTQ blocks.)
The assembly of mitochondrion is easy and could be done with many tools. we use Abyss that happens to be available as a module on Puhti.
First, we extract the reads mapped to the mitochondrion-like contig:
$ module load samtools
$ module load bedtools
$ mkdir -p mito/abyss
$ samtools view -h bams/sample-1_bwa.bam deg7180000006464 | samtools sort -n - \
| bedtools bamtofastq -i - -fq mito/abyss/sample-1_1.fq -fq2 mito/abyss/sample-1_2.fq
$ less mito/abyss/sample-1_1.fqThe trick is to use samtools sort -n to re-sort the reads such that the read-pairs are next to each other and can be directed to separate files. In fact, this assembly task could probably be done equally well with unpaired reads, but some other tasks may require pairing and it is good to be aware of the possibility.
With that, we can run Abyss. The parameters here are not optimised but seem to work for the task:
$ module load abyss
$ cd mito/abyss/
$ abyss-pe name=mitochondrion k=90 B=5M in="sample-1_1.fq sample-1_2.fq"
$ less mitochondrion-scaffolds.faThe second word in the header is the length and seems to be of the right magnitude. We can thus give it a more informative name and copy to the parent directory:
$ ( echo ">MT_ninespine" && tail -n +2 mitochondrion-scaffolds.fa ) > ../ninespine_MT.fa
$ cd ..As we now have re-assembled the mitochondrion and the sequence has changed, the original gene annotation cannot be correct. Gene models can be predicted de novo from RNA-seq data but there are also several ways of transferring gene annotations from a closely-related model species.
Ensembl data
Everyone working with genome data should be aware of Ensembl. On its home page, it is described as:
Ensembl is a genome browser for vertebrate genomes that supports research in comparative genomics, evolution, sequence variation and transcriptional regulation. Ensembl annotate genes, computes multiple alignments, predicts regulatory function and collects disease data.
The main Ensembl site emphasises the human genome and closely-related species helping understanding it through comparative analyses and contains the genomes of a couple of hundreds of vertebrate species. If no relatives of a study organism is among those, it’s worth checking also the sites for Rapid Release, Ensembl Bacteria, Ensembl Fungi, Ensembl Plants, Ensembl Protists, and Ensembl Metazoa.
The three-spined stickleback, Gasterosteus aculeatus, is one of the main Ensembl species and is used here.
BioMart and REST API
The Ensembl data can be browsed through the graphical genome browse and much of the data visualised in each screen can be downloaded with a few mouse clicks. On this course, we rather do it in large quantities and using programmable approaches.
Large quantities of data can be downloaded through a graphical interface at Ensembl BioMart. The Mart has also programmable interfaces e.g. through an R library (“biomaRt”).
Ensembl provides a REST API similar to that described in EEB-021. Unfortunately, the REST API only provides the latest release of Ensembl (it is updated every six months) and that has a version of stickleback genome without the mitochondrion. We therefore cannot use the API here although it is otherwise highly recommended and easy to use in Bash scripts through wget or curl.
FTP
We use the “Stickleback (BROAD S1)” genome from the Ensembl release 111. Find “Download FASTA” and “Download GFF3”, browse the FTP directories behind these links and find the files specified below. Get the full URL addresses of the files and insert the missing parts in the commands below:
$ cd mito
$ wget [fix the path]/Gasterosteus_aculeatus.BROADS1.dna.group.MT.fa.gz
$ wget [fix the path]/Gasterosteus_aculeatus.BROADS1.111.group.MT.gff3.gzWe can check that the files look like they should:
$ less Gasterosteus_aculeatus.BROADS1.dna.group.MT.fa.gz
$ less Gasterosteus_aculeatus.BROADS1.111.group.MT.gff3.gzAs the FASTA file is not compressed with bgzip and some programs do not accept compressed files at all, we decompress it first:
$ zcat Gasterosteus_aculeatus.BROADS1.dna.group.MT.fa.gz > threespine_MT.fa
$ samtools faidx threespine_MT.faAnnotation liftover
Below, we go through three approaches for transferring annotations from the three-spined stickleback mitochondrion to the nine-spined stickleback mitochondrion.
Liftover chain
The first approach uses “liftover chain”, a simplified representation of the homologous regions in two genomes. We build the liftover chain using the nextflow LiftOver pipeline. That pipeline uses minimap2 to align the two genomes and then computed the matching areas from that output. “nf-LO” is currently not provided at Puhti and it requires some helper tools that are also missing. Those have been installed at /projappl/project_2000598/bin and that directory has to be added to the path that the system uses to find the programs. Note that the “pipeline” itself doesn’t need to be installed but nextflow automatically retrieves it from GitHub.
With those, the pipeline can be run as:
$ module load nextflow
$ module load minimap2
$ PATH=/projappl/project_2000598/bin:$PATH
$ nextflow run evotools/nf-LO --source threespine_MT.fa --target ninespine_MT.fa --distance far --aligner minimap2 --outdir liftover_3sp_9sp --no_netsyntNote that the pipeline cannot be run in a login node as it requires too much memory. You can start an interactive SLURM session with the command sinteractive; the default settings are fine.
The resulting chain file is simple:
$ less liftover_3sp_9sp/chainnet/liftover.chainNote that the mitochondrion is circular and has no specified start and end position. The output from Abyss may not deterministic and it is possible that your ninespine_MT.fa has the break in a different position and the chain file is thus different.
The chain format is explained at the UCSC site.
CrossMap
The same “nf-LO” pipeline could be used to transfer coordinates from one genome to another but internally the pipeline uses the program CrossMap and it is much easier to use that directly. Load the module and check how the program works:
$ module load crossmap
$ CrossMap.py
$ CrossMap.py gff
$ CrossMap.py bedA series limitation of CrossMap is that it requires all the GFF elements to retain their length exactly as they were in the original file. This is unrealistic for many gene elements and many of the elements are left unmapped. There is no such requirement for the BED format and a solution would be to convert the GFF data to a multicolumn BED format (there are many different versions of BED for different purposes). I haven’t found a satisfactory method for that and wrote a simple script that packs the extra information in the GFF columns as the fourth column of a BED file; this can then be unpacked and converted back to GFF format.
$ gff_to_bed4=/projappl/project_2000598/bin/gff_to_bed4
$ bed4_to_gff=/projappl/project_2000598/bin/bed4_to_gff
$ zcat Gasterosteus_aculeatus.BROADS1.111.group.MT.gff3.gz | $gff_to_bed4 > Gasterosteus_aculeatus.BROADS1.111.group.MT.bedWith that, the liftover is then done as:
$ CrossMap.py bed liftover_3sp_9sp/chainnet/liftover.chain Gasterosteus_aculeatus.BROADS1.111.group.MT.bed ninespine_genes_crossmap.bedWe can see how the successfully transferred coordinates look and which annotated elements failed to be transferred:
$ less -S ninespine_genes_crossmap.bed
$ less -S ninespine_genes_crossmap.bed.unmapThe BED file can then be converted back to GFF:
$ cat ninespine_genes_crossmap.bed | $bed4_to_gff > ninespine_genes_crossmap.gff
$ less -S ninespine_genes_crossmap.gffBased on the observations, we can add a few more commands:
$ cat ninespine_genes_crossmap.bed | $bed4_to_gff | sort -k4,5n | awk '$3!="region"' > ninespine_genes_crossmap.gff
$ less -S ninespine_genes_crossmap.gffLiftOff
LiftOff doesn’t use a chain file but relies on pairwise alignments of genomic regions containing genes. The idea is that LiftOff uses the threespine gene annotations to extract regions from the threespine genome and then aligns these sequences against the ninespine genome. Using the alignments of full gene regions, it then calculates the positions of exons within the ninespine genome, creating gene annotations. The concept works nicely for long genomic elements (such as genes) but not for short elements (e.g. binding sites) or single bases.
LifOff is not provided at Puhti and we use a local installation:
$ module load minimap2
$ zcat Gasterosteus_aculeatus.BROADS1.dna.group.MT.fa.gz > threespine_MT.fa
$ liftoff=/projappl/project_2000598/bin/liftoff
$ $liftoff -g Gasterosteus_aculeatus.BROADS1.111.group.MT.gff3.gz -o ninespine_genes_liftoff.gff ninespine_MT.fa threespine_MT.faWe can again see how the successfully transferred coordinates look and which annotated elements failed to be transferred:
$ less -S ninespine_genes_liftoff.gff
$ less -S unmapped_features.txtMiniprot
As the third approach, we use miniprot. The program is one of many methods developed by Heng Li and conceptually similar to `minimap2´ but does the alignments between protein sequences and genomic sequences. For that, we need the threespine protein sequences that we download and recompress such that they can be indexed:
$ wget https://ftp.ensembl.org/pub/release-111/fasta/gasterosteus_aculeatus/pep/Gasterosteus_aculeatus.BROADS1.pep.all.fa.gz
$ zcat Gasterosteus_aculeatus.BROADS1.pep.all.fa.gz | bgzip -c > threespine.all.pep.gz
$ samtools faidx threespine.all.pep.gzWe then extract the IDs of mitochondrial protein-coding coding genes in the threespine annotations and pick them from the protein sequence file:
$ zcat Gasterosteus_aculeatus.BROADS1.111.group.MT.gff3.gz | grep ENSGACP | cut -d";" -f3 | cut -d"=" -f2 > threespine.MT.pep.txt
$ cat threespine.MT.pep.txt | xargs -n1 -I% samtools faidx threespine.all.pep.gz %.1 > threespine.MT.pepWith that, we can run miniprot:
$ miniprot=/projappl/project_2000598/bin/miniprot
$ $miniprot -T2 --gff ninespine_MT.fa threespine.MT.pep > ninespine_genes_miniprot.gffand see how the successfully transferred coordinates look :
$ less -S ninespine_genes_miniprot.gffThe option -T allows setting the genetic code for DNA translation. The numbers refer to those at the NCBI system.
We can summarise the number of elements in each annotation with the command:
$ ls ninespine_genes_*.gff | xargs -n1 -I% sh -c 'echo %; grep -v "#" % | cut -f3 | sort | uniq -c'Visualisation of gene models
The gene models from the three different methods can be compared visually using the R package Gviz. Gviz is an amazing package and allows making really stunning figures of genomic data. It’s worth browsing through the documentation page!
The following R code plots the genes in three panels:
setwd(paste0("/scratch/project_2000598/",Sys.getenv("USER")))
library(Gviz)
library(GenomicFeatures)
options(ucscChromosomeNames=FALSE)
axisTrack <- GenomeAxisTrack( )
txdb.cm <- makeTxDbFromGFF("mito/ninespine_genes_crossmap.gff")
cmTrack <- GeneRegionTrack(txdb.cm, chromosome="MT_ninespine", from=1, to=16840, showId=T,
name="CrossMap",fill="orange1",col=NA, transcriptAnnotation = NA)
txdb.lo <- makeTxDbFromGFF("mito/ninespine_genes_liftoff.gff")
loTrack <- GeneRegionTrack(txdb.lo, chromosome="MT_ninespine", from=1, to=16840, showId=T,
name="LiftOff",fill="orange1",col=NA, transcriptAnnotation = NA)
txdb.mp <- makeTxDbFromGFF("mito/ninespine_genes_miniprot.gff")
mpTrack <- GeneRegionTrack(txdb.mp, chromosome="MT_ninespine", from=1, to=16840, showId=T,
name="Miniprot",fill="orange1",col=NA, transcriptAnnotation = NA)
plotTracks(list(axisTrack, cmTrack, loTrack, mpTrack),showTitle = FALSE,transcriptAnnotation = "symbol",showTitle=T)It is easy to see that the main difference is the first panel containing many more elements. By default, LiftOff only consider “gene” fields, and miniprot, naturally, only considers protein-coding genes. The original annotation also include the RNA genes.
The annotated regions can also be compared with bedtools. First, it’s useful to check how the program is used:
$ bedtools
$ bedtools coverageand then try to compare the overlap of two sets:
$ bedtools coverage -a ninespine_genes_liftoff.gff -b ninespine_genes_miniprot.gff | less -SThe comparison across all predicted elements isn’t very useful as a CDS overlapping – in addition to the homologous CDS – with a mRNA and a gene isn’t very informative. (See the illustration at the web site.) It thus makes sense to consider one class of elements at a time.
Selecting “gene” reveals that Miniprot doesn’t report “gene” elements as the proteins come from “mRNA” and the program has no knowledge if multiple mRNAs come from the same gene. A comparison of mRNAs:
$ bedtools coverage -a <(awk '$3=="mRNA"' ninespine_genes_liftoff.gff) -b <(awk '$3=="mRNA"' ninespine_genes_miniprot.gff) | cut -f-5 | sort -k4n
$ bedtools coverage -b <(awk '$3=="mRNA"' ninespine_genes_liftoff.gff) -a <(awk '$3=="mRNA"' ninespine_genes_miniprot.gff) | cut -f-5 | sort -k4n
$ awk '$3=="mRNA"' ninespine_genes_[lm]*.gff | cut -f-5 | sort -k4n | column -tshows that, when the model exists, the differences are small. However, LiftOff has produced much fewer gene models.
The three approaches produce different results. Which of the methods matches each of the track (indicated on the left) in the figure?
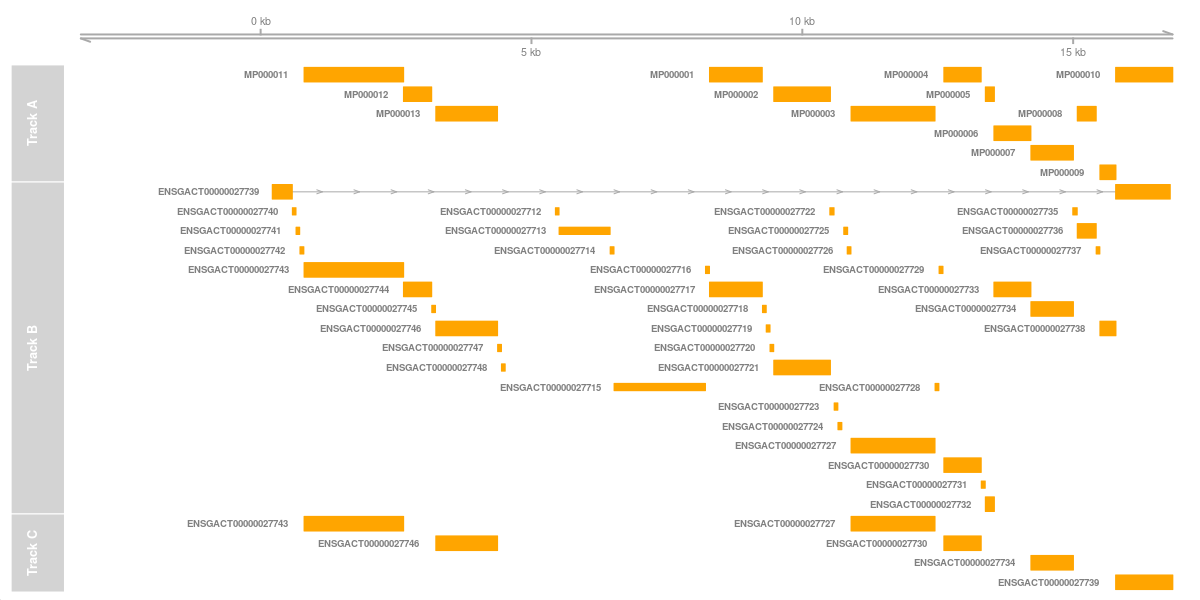
Exercise 3 in Moodle.
vulgar2gff3
As a fourth method, we can quickly test an approach that I developed before Miniprot was released. The idea is similar and builds on the Exonerate alignment program that can align protein sequences against genomic sequence.
My tool can be downloaded from Github:
$ module load git
$ git clone https://github.com/ariloytynoja/vulgar2gff3.git
$ less vulgar2gff3/vulgar2gff3.plThe beginning of the script reveals that the tool expects the DNA and protein sequence files to be of format “(+.nuc)” “(+.pep)”, that is consist of alphanumeric characters with suffixes “.nuc” and “.pep”. The code could be modified but now it’s simpler just to make copies of the files with the correctly formatted names:
$ cp ninespine_MT.fa nsp.nuc
$ cp threespine.MT.pep tsp.pepThe gene models are then predicted as:
$ module load exonerate
$ module load bioperl
$ exonerate -m p2g -t nsp.nuc -q tsp.pep --geneticcode 2 | perl vulgar2gff3/vulgar2gff3.pl > ninespine_genes_exonerate.gffThe behaviour of the script can be understood by looking at the output of exonerate:
$ exonerate -m p2g -t nsp.nuc -q tsp.pep | less -SThe key information are the lines starting with “vulgar:”
$ exonerate -m p2g -t nsp.nuc -q tsp.pep --showalignment F “VULGAR” is very similar to “CIGAR” discussed earlier and describes the alignment when the two sequences are known.
A comparison of the two protein-sequence-based approaches shows that the results are highly similar:
$ awk '$3=="mRNA"' ninespine_genes_[em]*.gff | cut -f-5 | sort -k4n | column -tHowever, exonerate appears to find one gene missed by miniprot. Secondly, my script “packages” each “mRNA” element to a “gene” of its own and produces GFF files compatible with the downstream methods requiring that.
Variant calling
In addition to gene annotations, also the variant calling is now erroneous. Given the skills learned previously, it is relatively straightforward to recall the mitochondrion only.
First, we need to prepare the new reference, ninespine_MT.fa:
$ WORK=/scratch/project_2000598
$ cd $WORK/$USER
$ module load bwa
$ module load samtools
$ bwa index mito/ninespine_MT.fa
$ samtools dict mito/ninespine_MT.fa > mito/ninespine_MT.dict
$ mkdir mito/fastq mito/bams mito/gvcfs mito/vcfsThen, we can write a script that handles a sample from the start to the end. Note that the very last command has an additional parameter -ploidy 1 to tell that the mitochondrion is haploid and should be called as such:
$ cat > extract_map_and_call.sh << 'EOF'
#!/bin/bash
#SBATCH --time=15:00
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=2
#SBATCH --mem=6G
#SBATCH --partition=test
#SBATCH --account=project_2000598
#SBATCH --output=logs/extract_map_and_call-%j.out
gatk3=/projappl/project_2000598/bin/gatk3
ref=mito/ninespine_MT.fa
fastq=mito/fastq
bams=mito/bams
gvcfs=mito/gvcfs
module load bwa
module load gatk
module load samtools
module load bedtools
inbam=$1
smp=$(basename $inbam _mdup.bam)
samtools view -h $inbam deg7180000006464 | samtools sort -n - \
| bedtools bamtofastq -i - -fq $fastq/${smp}_1.fq -fq2 $fastq/${smp}_2.fq
rgroup="@RG\tID:$smp\tLB:Lib\tSM:$smp\tPL:Plfm"
bwa mem -t2 -R ${rgroup} ${ref} $fastq/${smp}_1.fq $fastq/${smp}_2.fq \
| samtools fixmate -m - - | samtools sort - -o $bams/${smp}_bwa.bam \
&& samtools index $bams/${smp}_bwa.bam
$gatk3 -T RealignerTargetCreator \
-R ${ref} -I $bams/${smp}_bwa.bam -o $bams/${smp}.intervals
$gatk3 -T IndelRealigner \
-R ${ref} -I $bams/${smp}_bwa.bam -targetIntervals $bams/${smp}.intervals -o $bams/${smp}_real.bam
samtools markdup $bams/${smp}_real.bam $bams/${smp}_mdup.bam
samtools index $bams/${smp}_mdup.bam
gatk HaplotypeCaller \
-R ${ref} -I $bams/${smp}_mdup.bam -O $gvcfs/${smp}.gvcf.gz -ERC GVCF -ploidy 1
EOFWe can now process our sample:
$ sbatch extract_map_and_call.sh bams/sample-1_mdup.bamand follow its progress in the queue:
$ squeue --meand in the log file:
$ tail logs/extract_map_and_call-*.outOnce it has finished, we can see the new GVCF file:
$ less -S mito/gvcfs/sample-1.gvcf.gzWe have previously used gatk GenotypeGVCFs to call GVCF files of many samples but it can also be applied to a simple sample:
$ module load gatk
$ gatk GenotypeGVCFs -R mito/ninespine_MT.fa -V mito/gvcfs/sample-1.gvcf.gz -O mito/vcfs/sample-1.vcf.gz
$ bcftools query -f '%CHROM\t%POS\t%REF\t%ALT[\t%SAMPLE=%GT]\n' mito/vcfs/sample-1.vcf.gzUnsurprisingly, there are no variants as the reference is based on the very same individual.
However, if the same is done for another sample:
sbatch extract_map_and_call.sh $WORK/shared/bams/sample-14_mdup.bamthe resulting VCF looks more informative.
Variant annotation
We have learned the process of variant calling earlier and it is not repeated here. Instead, we use a ready-made VCF file:
$ cp $WORK/shared/mito/100_samples_MT.vcf.gz mito/
$ bcftools query -f '%CHROM\t%POS\t%REF\t%ALT[\t%GT]\n' mito/100_samples_MT.vcf.gz | less -S
$ bcftools query -f '%CHROM\t%POS\t%REF\t%ALT\t%INFO/AF\n' mito/100_samples_MT.vcf.gz | less -SThere are many tools for annotating variants based on GFF file, one of the easiest being bcftools csq:
$ bcftools csqWe can do that for our mitochondrial data and explore the output:
$ bcftools csq -f mito/ninespine_MT.fa -g mito/ninespine_genes_exonerate.gff mito/100_samples_MT.vcf.gz -Oz -o mito/100_samples_MT_exonerate.vcf.gz
$ bcftools query -f '%CHROM\t%POS\t%REF\t%ALT\t%INFO/BCSQ\n' mito/100_samples_MT_exonerate.vcf.gz | less -SThe list looks impressive but is probably incorrect: the amino acid changes were inferred based on the standard genetic code, not the mitochondrial one. I’m not aware of an option in bcftools csq for the annotation of mtDNA VCF variants.
SnpEff/SnpSift
A significantly more complex (and comprehensive) method for variant annotation is the SnpEff/SnpSift package. Unfortunately I have no personal experience on this method and can only provide a preliminary introduction.
SnpEff is a Java program and requires the config file to be in certain position relative to the program file. To get that right, we copy the programs and the default config file to a local directory:
$ cd $WORK/$USER
$ mkdir -p mito/snpeff/data/nsp_mt
$ cp $WORK/shared/snpeff/* mito/snpeffSnpEff can download the relevant data for a huge number of reference genomes from different databases. The genomes supported by the program can be seen in the config file (there’s lots of them!):
$ cd mito/snpeff
$ less -S snpEff.configOurs is naturally not there as it was generated above and we have build a database from the existing files.
SnpEff assumes the input files to have standardised names and be placed in a subdirectory under data; data can contain multiple genomes and the name of the subdirectory is used in the commands to specify which genome is used. Above, we created subdirectory nsp_mt. We have to copy the reference genome (here, for the mitochondrion) and its annotation file using the standardised names:
$ cp ../ninespine_MT.fa data/nsp_mt/sequences.fa
$ cp ../ninespine_genes_crossmap.gff data/nsp_mt/genes.gffWe then have to add the information about the new genome in the config file. As the codon table is not the standard one, we have to specify that as well:
$ echo "nsp_mt.genome : nsp_mt" >> snpEff.config
$ echo "nsp_mt.MT_ninespine.codonTable : Vertebrate_Mitochondrial" >> snpEff.configWe can then load the Java module and run the program:
$ module load biojava/21
$ module load samtools
$ java -jar snpEff.jar build -gff3 -v nsp_mt -noCheckCds -noCheckProtein
$ ls -lh data/nsp_mt/This builds a database for the target genome. We can then annotate our VCF file; here the output is fed into bcftools view to compress it immediately:
$ java -jar snpEff.jar -v -stats 100_samples_MT_snpEff_crossmap.html nsp_mt ../100_samples_MT.vcf.gz | bcftools view -Oz -o 100_samples_MT_snpEff_crossmap.vcf.gzIn addition to annotating the variants in the output file, the command above creates a HTML file, called “100_samples_MT_snpEff_crossmap.html”, with a summary of the annotations. This HTML file can be opened with Jupyter.
For comparison, we can re-build the annotation database using the GFF file generated with Exonerate. If the process would be more time-consuming, it would make sense to place them in different subdirectories. Here, we just rerun the database build and generate another HTML file and annotated VCF file::
$ cp ../ninespine_genes_exonerate.gff data/nsp_mt/genes.gff
$ java -jar snpEff.jar build -gff3 -v nsp_mt -noCheckCds -noCheckProtein
$ java -jar snpEff.jar -v -stats 100_samples_MT_snpEff_exonerate.html nsp_mt ../100_samples_MT.vcf.gz | bcftools view -Oz -o 100_samples_MT_snpEff_exonerate.vcf.gzThe program SnpSift allows filtering and analysing the annotations but much of the functionality can also be done with bcftools. We can simply browse all the annotations:
$ bcftools query -f '%CHROM\t%POS\t%REF\t%ALT\t%INFO/ANN\n' 100_samples_MT_snpEff_crossmap.vcf.gz | less -Sor include filtering criteria in the bcftools query command:
$ bcftools query -i 'ANN~"MODERATE"' -f '%CHROM\t%POS\t%REF\t%ALT\t%INFO/ANN\n' 100_samples_MT_snpEff_exonerate.vcf.gz | less -S
$ bcftools query -i 'ANN~"HIGH"' -f '%CHROM\t%POS\t%REF\t%ALT\t%INFO/ANN\n' 100_samples_MT_snpEff_exonerate.vcf.gz | less -SAnnotation of codon positions
Instead of annotating the amino-acid changes that the variants cause, we can simply compute their positions within the codons. For that, we generate a simple awk script:
$ cat > gff2cpos.awk << 'EOF'
$3=="CDS" && $7=="+"{
OFS="\t";
pos=1;
if($8==1){pos=3}
if($8==2){pos=2}
for(i=$4;i<=$5;i+=1){
print $1,i,i,pos;
pos=pos+1;
if(pos>3){pos=1}
}
}
$3=="CDS" && $7=="-"{
OFS="\t";
phase=($5-$8-$4+1)%3;
pos=3;
if(phase!=0){pos=phase}
for(i=$4;i<=$5;i+=1){
print $1,i,i,pos;
pos=pos-1;
if(pos<1){pos=3}
}
}
EOFand then extract the first, second and third codon positions into seperate files:
$ awk -f gff2cpos.awk mito/ninespine_genes_exonerate.gff | sort -k2n | less
$ awk -f gff2cpos.awk mito/ninespine_genes_exonerate.gff | sort -k2n | grep 1$ > mito/codon1.tab
$ awk -f gff2cpos.awk mito/ninespine_genes_exonerate.gff | sort -k2n | grep 2$ > mito/codon2.tab
$ awk -f gff2cpos.awk mito/ninespine_genes_exonerate.gff | sort -k2n | grep 3$ > mito/codon3.tabWith those, we can now extract the variants at different codon positions
$ bcftools view -T mito/codon1.tab mito/100_samples_MT.vcf.gz \
| bcftools query -f '%CHROM\t%POS\t%REF\t%ALT\t%INFO/AF\n' | less
$ for i in 1 2 3; do
echo -n "codon$i: "
bcftools view -H -T mito/codon$i.tab mito/100_samples_MT.vcf.gz| wc -l
doneor those within genes or outside of them:
$ cut -f1,4,5 mito/ninespine_genes_crossmap.gff | sort -k2n | bedtools merge -i - > mito/ninespine_genes_crossmap.tab
$ bcftools view -Tmito/ninespine_genes_crossmap.tab -H mito/100_samples_MT.vcf.gz | wc -l
$ bcftools view -T^mito/ninespine_genes_crossmap.tab -H mito/100_samples_MT.vcf.gz | wc -lWe can see that only 7.49% of the mitochondrion is not covered by any annotations in that file, allowing to compare whether the variants are enriched inside or outside of them:
$ bedtools genomecov -i mito/ninespine_genes_crossmap.gff -g mito/ninespine_MT.fa.fai | head -1MT_ninespine 0 1261 16840 0.0748812One should note, though, that this includes both protein-coding genes and RNA genes. 32.2% of the mitochondrion is not covered by a protein-coding exon (CDS):
$ bedtools genomecov -i <(grep CDS mito/ninespine_genes_crossmap.gff) -g mito/ninespine_MT.fa.fai | head -1MT_ninespine 0 5429 16840 0.322387The allele counts were computed for the three codon positions (replace “X” with numbers 1, 2 and 3 in the code below) like this:
bcftools view -m2 -M2 -v snps -T codonX.tab 100_samples_MT.vcf.gz | vcftools --vcf - --counts2 --out codonXand the results were plotted like this:
library(ggplot2)
library(dplyr)
# copy the block of code below and replace "X" with 1, 2 and 3
datX <- read.table("codonX.frq.count",skip=1,col.names = cn)
frqX <- datX %>% mutate(FR=pmin(FD,100-FD)) %>% count(FR) %>% mutate(POS="codonX",N=sum(n))
# running the code above three times (with X->1, X->2 and X->3) produces
# frq1, frq2 and frq3; combine and plot the counts
frq <- rbind(frq1,frq2,frq3)
ggplot(frq) + geom_col(aes(x = FR, y = n/N, fill = POS),position = "dodge")Replace “X” with 1, 2 and 3 and complete the analysis using the skills learned in the previous exercises. In the SFS plot below, which colour represents each codon position?
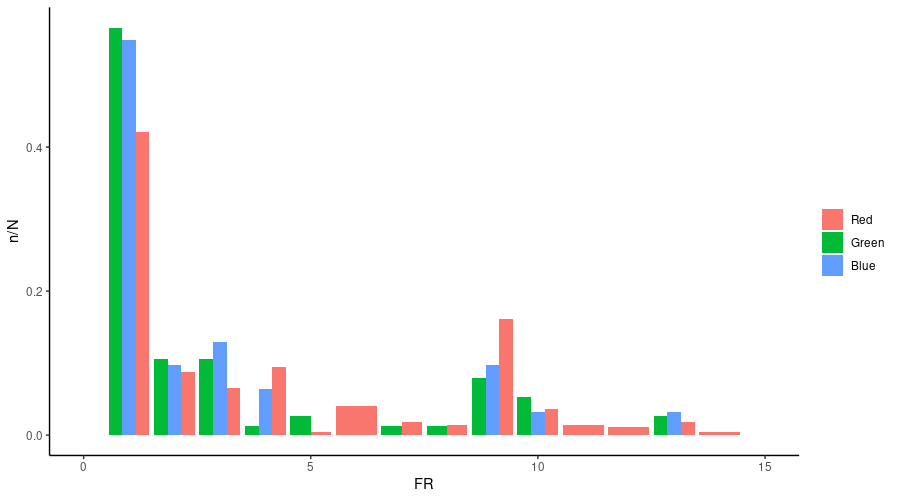
Exercise 6 in Moodle.
The BAM files contain all the information that was in the original FASTQ reads. Reads of a specific region can be extracted and reutilised. De novogene annotation can be a demanding task but often gene annotation information can be transferred from a closely-related species. Gene annotations can be used to annotate SNP and indel variants. Variant annotation is extremely sensitive to the quality of the gene models: if the reading frame of genes is wrong, the variant annotation is also wrong.
The quality of gene annotations should be checked more rigorously than done here. If the variant annotation is used to identify causative mutations, possible computational errors can be identified manually. In large-scale summary analyses (such as the comparison of codon positions across lots of genes), the gene models should be thoroughly tested.